17. Creating a new template
VEGA and VEGA ZZ uses two types of template files: the former is for atom types, and the latter is for atomic charges.
By ATDL (Atom Type Description Language), you can expand VEGA adding new atom types and/or new force field templates. Actually, VEGA supports the following pre-defined templates:
| Force Field | Package |
| AM1BCC | AM1BCC. |
| AMBER | Amber. |
| AUTODOCK | AutoDock 4 force field (based on AMBER). |
| BOND | Used by VEGA to calculate the bond types (single, double, partial double and triple). |
| BROTO | Broto and Moreau atom types for logP calculation. |
| CFF91 | Accelrys CFF91. |
| CHARMM | Accelrys Quanta/CHARMm. |
| CHARMM22_LIG | CHARMM 22 for ligands, including CHARMM22_PRO. |
| CHARMM22_LIPID | CHARMM 22 for lipids. |
| CHARMM22_NA | CHARMM 22 for nucleic acids. |
| CHARMM22_PRO | CHARMM 22 for proteins. |
| CHARMM27 | CHARMM 27 for proteins. |
| CHARMM36_GEN | CHARMM 36 for generic use. The use of this template is not recommended for proteins and nucleic acids. |
| CRIPPEN | Ghose and Crippen atom types for logP calculation. |
| CRIPPEN_MR | Ghose and Crippen atom types for molar refractivity calculation |
| CVFF | Accelrys CVFF. |
| GRID | Grid. |
| GROUPS | Used by VEGA to detect the functional groups. |
| HBOND | H-bond atom types (for internal use). |
| MENG | By Elanie C. Meng and Richard A. Lewis. |
| MM+ | MM+. |
| MM2 | MM2 by N .L. Allinger. |
| MM3 | MM3 by N .L. Allinger. |
| MMFF | MMFF94. |
| OPLS | OPLS. |
| SP4 | Used by VEGA to generate the AMMP input files. |
| TRIPOS | Sybyl by Tripos. |
| UNIV | Used by VEGA to assign the Gasteiger-Marsili atom charges. |
| VINA | AutoDock Vina force field (based on AMBER). |
A force field template is a file storing the atom type descriptions with uppercase name (corresponding to the force field name) and .tem lowercase extension (e.g. AMBER.tem, CVFF.tem, etc). All template files are placed in Data directory. Please remember that the .tem extension is for all VEGA templates and not for force field only.
In all template files the first column can contain special control characters:
| Character | Description |
| ; | Comment marker |
| # | Keyword or command marker |
The first line must contain a keyword needed for file type recognition. For force field it must be:
#TemplateFF [TEMPLATE_NAME] [VERSION]
where TEMPLATE_NAME is the name of the force field template and VERSION is the revision number.
#TemplateFF CVFF 3.0
After this keyword, you can place the atom type description. The first
column is the atom type name (max 8 characters), the second is the atom description in
ATDL and the third contains the description of bonded atoms (also in ATDL).
In this last column, each group of atoms limited by parenthesis contains all atoms bonded
to precedent atom:
C-300 (O-100 O-100)
This line describes a carboxylic carbon: a sp2 carbon bonded to two oxygens making one bond only. More than one levels of parenthesis can be used for complex description of atom types:
C-300 (O-100 O-200 (C-900) C-900)
This line describes a carbonylic carbon of an ester group, bonded to a
generic carbon. The O-200 is also bonded to a generic carbon.
Please remember that VEGA reads the line from left to right and thus the more restrictive
atom description must placed in more left side of line:
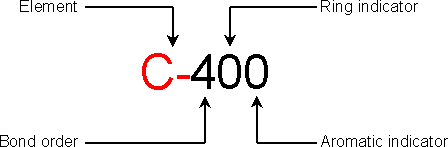
C-400 (C-300 X-900 X-900 X-900)
and not:
C-400 (X-900 X-900 C-300 X-900)
If VEGA finds a C-300 as first or second atom bonded to a sp3
carbon, this is recognized as a more generic X-900 atom and can't be reassigned to
the next more specific description.
The description sequence of each atom type goes from more to less specific, from upper to
lower line:
cn C-400 (N-300 X-900 X-900 X-900) ; more specific c C-400 (X-900 X-900 X-900 X-900) ; less specific
If the order of this two lines is swapped, when VEGA finds a carbon bonded to a sp3 nitrogen, the atom type recognized is a generic c an not a cn.
Each atom can be defined by a five character string. The first two characters are the element symbol of atom. If the element symbol is one character only, the second character must be a dash (-). For a better description, special elements can be used:
| Special element | Description |
| X | Any atom. |
| # | Heavy atom (all atoms excluding hydrogens). |
| $ | Any atom excluding carbons and hydrogens. |
| @ | Halogen (F, Cl, Br and I). |
The third character is the bond order: use values from 1 to 6 for real bond order,
0 for non-bonded atom and 9 for a bonded atom with a non-specified bond order.
The fourth character is the ring indicator: use values from 3 to 7 if the atom is a
3 to 7-ring member, 0 for a non-ring member atom and 9 for a non-specified ring atom.
The fifth character is the aromatic indicator: 0 for non-aromatic atom and 1 for
aromatic atom.
The ATDL language allows to use AND, OR and NOT operators (&, | and !) inside a
logical expression included between square.
Examples:
This template file is much different from the first one, because the atom recognition is based on the residue names and the atom names. The control characters are the same of the force field template.
The first line must contain a keyword needed for file type recognition. For force field it must be:
#TemplateCharge [TYPE] [TEMPLATE_NAME]
where TYPE is the template charge type (Gasteiger or
Fragments) and TEMPLATE_NAME is the name of the template. Please remember
that the template name must be the same one of the file without the extension.
Example:
#TemplateCharge Fragments CHARMM22_CHAR
After this keyword it could be present the optional template title/description:
#Title [TEMPLATE_TITLE]
Spaces and special characters are allowed.
Example:
#Title Gasteiger-Marsili charges
After these two keywords, the file can be different if the template type is Gasteiger or Fragments
17.2.1 Gasteiger template
The Gasteiger template is very easy: after the header it's a list of records one for each line. Each record has six fields as reported in the following table:
| Field | Description |
| Type | Atom type. See the UNIV.tem file in the Data directory. |
| a | Gasteiger a parameter. See Tetrahedron, 36, 3219, 1980 and Croat.Chem.Acta, 53, 601, 1980. |
| b | Gasteiger b parameter. |
| c | Gasteiger c parameter. |
| d | Gasteiger d parameter (a + b + c). |
| Charge | Formal charge. |
17.2.2 Fragment template
The fragment template is a little bit complex because it uses some keywords. To define a new residue, you must use the #ResName tag:
#ResName [NAME1] [NAME2] ... [NAME16]
e.g. #ResName ALA ALAN
In this way, you define a new residue that it could have one of the specified names. The maximum number of names is 16 and the maximum length of each name is 4 characters.
This tag could be followed by other optional keywords:
#Id [ID]
e.g. #Id AA_ALA
This command defines an unique residue identificator. It can be used by the #Call command (see below) and its maximum length is 31 characters.
#Description [SHORT_DESCRIPTION]
e.g. #Description Alanine (protonated N-terminus)
It allows to specify a short description for the residue or for the macro (see below). Its maximum size is 127 characters.
#Charge [CHARGE]
e.g. #Charge 1.0
This optional keyword specifies the residue total charge. The number should be a positive or a negative floating point number.
After these optional keywords, that must be after the #ResName tag, the atom section begins. Each atom is defined in a line with the following fields:
[CHARGE] [GROUPID] [BONDS] [NAME1] [NAME2] ... [NAME8]
Where:
| Field | Description |
| CHARGE | Atom partial charge. |
| GROUPID | Group/fragment identification number. It's a positive integer starting from 1 to 255. |
| BONDS | Number of atom bonds. It can be from 0 (non bonded) to 6. If it's greater than 6, the number of bonds isn't checked. |
| NAME1 ... NAME8 |
The atom names. The maximum number of atom names (aliases) is 8 and their maximum length is 4 characters. |
e.g. 0.3100 1 1 HN H H1
This is a complete residue template example:
#ResName ALA #Id ALA #Description Alanine #Charge 0.0000 -0.4700 1 3 N 0.3100 1 1 HN H H1 0.0700 1 4 CA 0.0900 1 1 HA -0.2700 2 4 CB 0.0900 2 1 HB1 0.0900 2 1 HB2 0.0900 2 1 HB3 0.5100 3 3 C -0.5100 3 1 O
In red are reported the optional keywords.
In order to simplify the template writing and to make more compact the file size, it's possible to create macros inside the file that must be defined before the use. To begin a new macros, you must use the following command:
#Define [MACROID]
e.g. #Define AMINO_CT
Where the MACROID is the unique identification name of the macro. It have the same function of the #Id command inside the #ResName section. Inside the macro, you can use the #Description, #Call, #Delete commands and the atom records.
#Call [RESIDUEID_OR_MACROID]
e.g. #Call AA_ALA
This command call a residue or a macro executing its commands. It can be placed inside a macro or a residue section.
#Delete [ATOM_NAME]
e.g. #Delete O
This keyword deletes an atom previously defined in a residue section.
Please remember that an atom record inside a macro could replace a previous one if they have the same first atom name. This is a macro example:
#Define AMINO_CT #Description C-terminus 0.3400 9 3 C -0.6700 9 1 OT1 OCT1 O1 O -0.6700 9 1 OT2 OCT2 O2 OXT #Delete O
Using this macro and an aminoacid residue definition, it's possible to obtain a new one specific for the C-terminal aminoacid:
#ResName ALA ALAC #Id ALAC #Description Alanine (negative C-terminal) #Charge -1.0000 #Call ALA #Call AMINO_CT
The first call copies the atom definitions from the ALA residue and the second call applies the AMINO_CT macro that change the C atom, add the two carboxyl oxygen, and delete the carbonyl oxygen (O).