6.15 Build a solvent cluster
By this dialog box, that can be shown
selecting Edit
![]() Build
Build
![]() Cluster menu item, it 's possible
to build a solvent cluster useful for the solute solvation (for more information,
click here). In order to create a new solvent
cluster usable by VEGA ZZ, you should follow these steps:
Cluster menu item, it 's possible
to build a solvent cluster useful for the solute solvation (for more information,
click here). In order to create a new solvent
cluster usable by VEGA ZZ, you should follow these steps:
6.15.1 The graphic interface
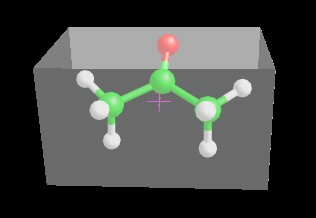
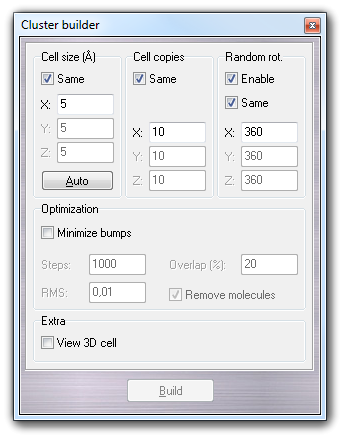
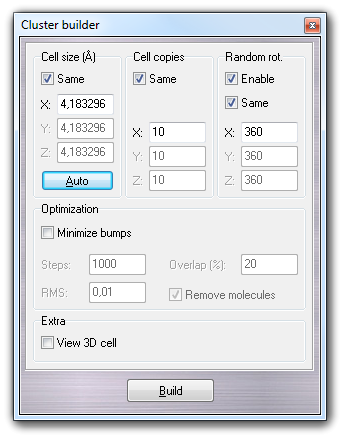
The following dialog box allows to build a cubic solvent cluster, specifying the dimensions of the box containing a single solvent molecule (Cell size) and the number of cell copies in X, Y, and Z directions (Cell copies).
|
Clicking Auto, the cell size is assigned automatically by the monomer
dimensions, considering the covalent radii of each atom. Clicking Build, the cluster is generated. The
parameters shown above allow to create an ordered cluster.
If you want to obtain a disordered system, you must check Enable in Random
rot. group-box, specifying the X, Y and Z ranges used to generate the
random rotation of each monomer placed in the cluster.
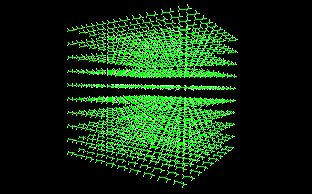
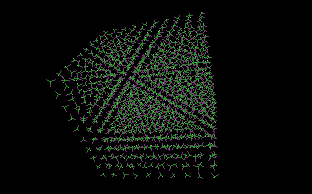
This is an application example: a chloroform cluster is built using random rotation with 360º rotation ranges (the same value for all three axis). |
|
|
If the cell size is too small, the bumping probability between close monomers is high. It's possible to reduce this problem, checking Minimize bumps in the Optimization group-box. The position and the rotation of each monomer placed in the cluster is minimized using the fast Hooke-Jeeves algorithm. You can indicate number of minimization Steps and the RMS. The Overlap field contains the value of the maximum atom overlapping between two near molecules. Enabling Remove molecules, the bumping molecules that can't be optimized, are automatically removed from the cluster.
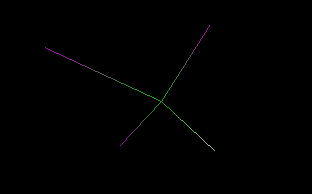
Clicking View 3D
cell, a transparent box is shown in order to highlight the cell dimension.
The magenta cross indicates the molecule symmetry center. You can change the
molecule orientation in the cell using the move molecule function that can be
enabled selecting Move
![]() Molecule item in the popup menu.
Molecule item in the popup menu.