6.11 Add ions
The capability to add the ions is based on the source code (SODIUM by Alexander Balaeff), developed by the Theoretical Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign.
6.11.1 Usage
Selecting Edit
![]() Add
Add
![]() Ions in the
main menu, you can add one or more counter ions to the active molecule:
Ions in the
main menu, you can add one or more counter ions to the active molecule:
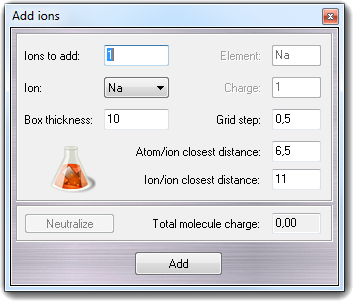
You can indicate the number of ions (Ions
to add), the Ion element, the Box thickness surrounding the macromolecule, the
Grid step to build the grid used to
place the ions, the Atom-ion closest distance and the Ion/ion closest
distance
in order to reduce the electrostatic repulsion. More in details, the Box
thickness parameter is the thickness of the grid margin, surrounding the
macromolecule (in Ångstroms): as an example, imagine a molecule with a cubic
shape of 20x20x20 Å. A 10 Å Box thickness means that the ions will be
placed in a theoretical box of 40x40x40 Å defining a 10 Å margin around the
molecule. If you select Custom as
ion type, you can specify other non-predefined ions typing the Element and the
Charge in the two fields placed at top right corner of the window. If the
molecule charge is a multiple of the ion charge, you can press the Neutralize
button to calculate the number of ions needed to neutralize the system.
Click Add button to calculate the position of all ions: be
patient, because the calculation is time expensive. The calculation progress is
shown by a graphic bar.
WARNING:
If the molecule doesn't have atomic charges, you can't add the ions because
isn't possible calculate the electrostatic interactions. In this case, VEGA
shows a requester to add the atomic charges.
6.11.2 About the method
This program places the required number of ions around a system of electric charges, e.g., the atoms of a molecule. The ions are placed in the nodes of a cubic grid, in which the electrostatic energy achieves the smallest values. The energy is re-computed after placement of each ion. A simple Coulombic formula is used for the energy:
Energy(R) = Sum(i_atoms,ions) Q_i / |R-R_i|
All the constants are dropped out from this formula, resulting in some
weird energy units; that doesn't matter for the purpose of energy comparison.
To speed the program up, the atoms of the macromolecule are re-located to the grid nodes, closest to their original locations. The resulting error
is believed to be minor, compared to that resulting from the one-by-one ions placement, or from using the simplified energy function.