9.2 Database explorer
The Database explorer is a tool
to manage molecular databases, allowing some basic operations that can be
classified in two main groups: the database operations
(open, synchronize, close and close all) and molecule management
functions (find, get, put, remove, rename and update). These functions are
accessible by buttons and popup menus.
The Database explorer is shown automatically when a database is opened
by Open/create file requester or selecting File
![]() Database
Database
![]() Explorer
in main menu. Its window is full resizable
and the Database and Molecule boxes are resizable inside the
window also.
Explorer
in main menu. Its window is full resizable
and the Database and Molecule boxes are resizable inside the
window also.
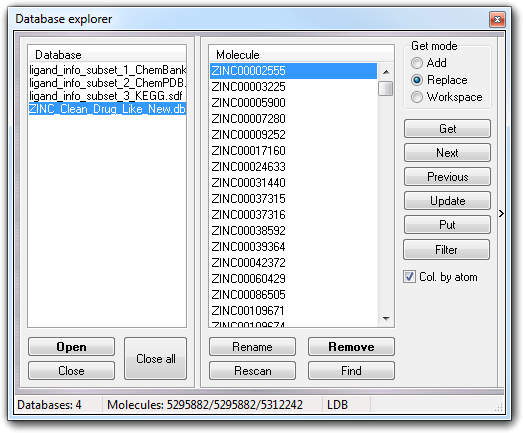
The left box shows the gadgets to manage the database: Database list to select the current database, Open button to open/create a database, Close button to close the selected database and Close all button to close all databases. The user can also open the database by drag & drop operations: drop one or more database files over the Database box to open them. Clicking with the right mouse button on the database items, a popup menu is shown: it replicates the button functions (Open, Close and Close all) and includes the Synchronize item that allows to synchronize the large databases (LDB, see below).
The right box includes all controls to manage the molecules. To extract a structure from the database, you must select one or more molecules in Molecule list (multiple selections are allowed), choose Get mode to add/replace the molecule in the current workspace or to place the molecule in a new workspace, and finally click Get button. To do the same operation, you can double click the molecule name or use the popup menu or hit the return key. Next and Previous buttons are useful to scan the database, because allow to get sequentially the structures from the database.
Expanding the window, clicking the vertical button on the right side ( > ), the 2D structure is shown before the extraction from the database.
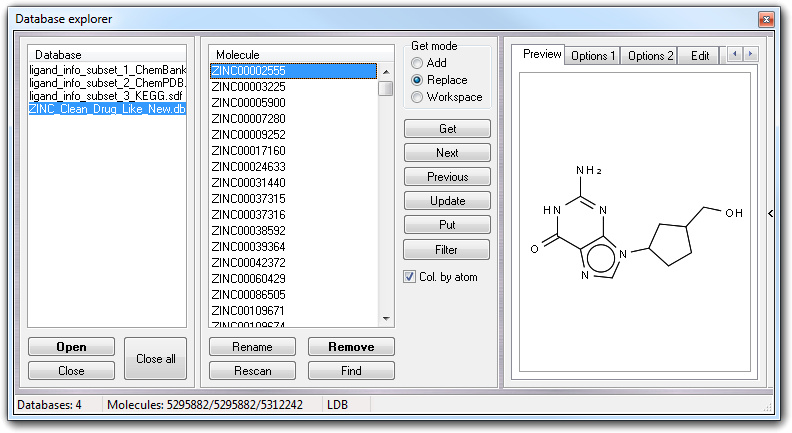
Showing the context menu of the preview area, you can copy the 2D sketch to the clipboard in vector format, or print. The 2D preview is not available if the database doesn't include the SMILES field or is not in SDF format.
9.2.1.4 Inserting molecules into the database
To insert a molecule into the database, you must open its structure in the current
workspace, thus click Put button: a dialog box is shown to edit the
molecule name. Clicking Ok button, the molecule
is added to the database using the default parameters (molecule format,
compression, connectivity and constraints) defined when the database is created.
Alternatively, you can drag & drop one or more molecule files on the
Molecule box to add them without opening in the current workspace. Moreover,
it's
possible to copy one or more molecules from a database to another one, just
dragging & dropping the molecules from the Molecule box to a previously
opened database shown in Database box. Dragging & dropping a database over
another one, all molecules are copied to the destination database. During the
copy operation, the progress bar is shown at the bottom of VEGA ZZ main
window. If you want to stop this operation, you can click Abort button
at the right of the progress bar.
When you insert or update molecules, they can activate the pre-processing
functions that allow to:
To enable/disable or set the parameters of these features, you must expand the Database explorer window, clicking the slim > button on the right of the window.
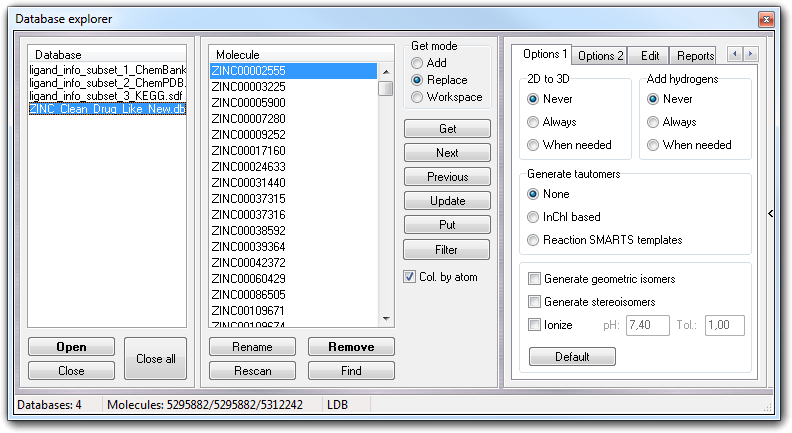
In Option 1 tab, you control the 2D to 3D conversion (2D to 3D
box): you can disable (Never), enable (Always), enable if needed (When
needed) this feature. In last case, the conversion to 3D is performed
only if the starting structure is 2D and if the molecule is already in 3D, the
conversion is skipped. In the 2D to 3D conversion, is strongly recommended to
perform the energy minimization.
Add hydrogens box allows to disable (Never), enable (Always),
enable if needed (When needed) the addition of missing hydrogens. When
needed adds the hydrogens only if they are really missing. The best algorithm is
automatically selected on the basis of molecule type (protein, nucleic acid
and generic organic molecule). For more information about the method to add the
hydrogens, click here.
In Generate tautomers box, you can disable (None) or enable the tautomer generation with two different algorithms: 1) based on the InChI string (InChI based option); 2) based on reaction templates (Reaction SMARTS templates option).
Checking Generate geometric isomers and/or Generate stereoisomers, you can activate the automatic build respectively of all possible geometric- and stereo- isomers.
If you have to change the ionization state of the molecule, you must check Ionize, thus you can specify the pH at which you want to ionize the molecule and the tolerance (Tol., in pH units) used to establish which protonation state is largely present (for more information, click here).
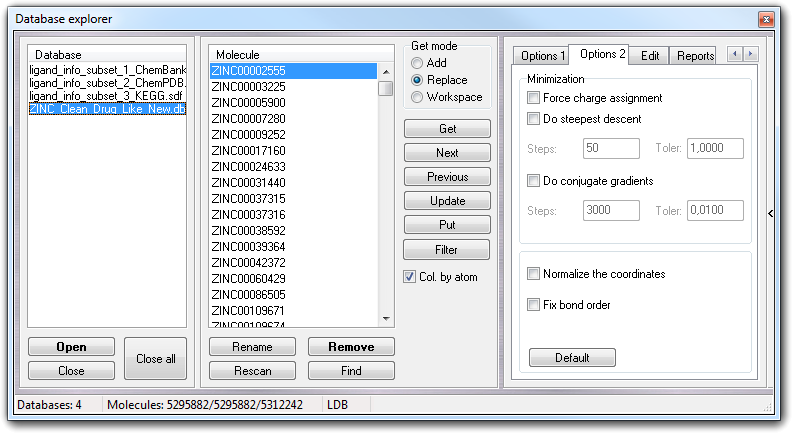
In Options 2 tab, you can find the Minimization box in which you can control the parameters for the energy minimization phase: checking Do steepest descent, the steepest descent minimization is performed for the specified number of Steps or until the gradient is not satisfied (Toler value). In the same way, clicking Do conjugate gradients, you can enable the conjugate gradients minimization that will be stopped satisfying the number of Steps or the gradient (Toler). Checking Remove counterions & waters, the counterions and water molecules are removed, while checking Keep only the largest molecule, if the workspace includes more than one molecule, only the largest one is kept. The size of the molecule is calculated counting the number of heavy atoms. Moreover, if the atomic charges are uncertain, you can force their assignment checking Force charge assignment.
Clicking Normalize the coordinates, the molecule
is translated at the origin of the Cartesian axis. Clicking Fix bond order,
the bond types are changed by valence saturation (e.g. the alternate
single-double bonds in aromatic rings are converted to partial double bonds).
If you need to revert to default parameters, click Default
button.
When you put a molecule in the database, some molecular descriptors are calculated and it's possible to view and edit them by Edit tab.
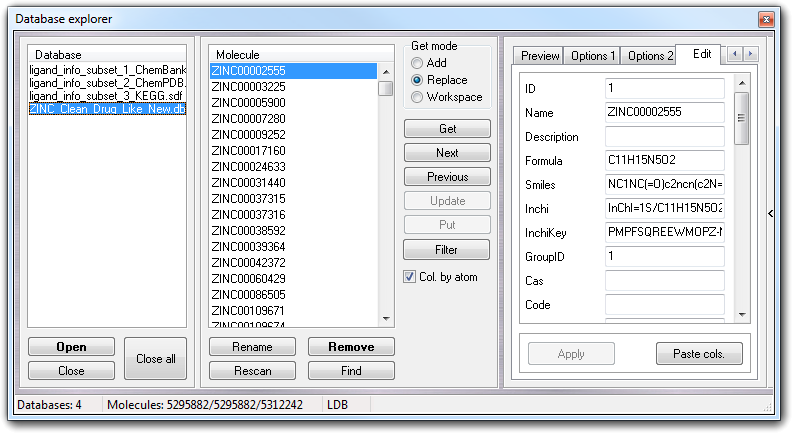
To apply the changes, just press return key or click Apply button. VEGA ZZ can manage user-defined fields (e.g. activity, experimental data, etc), but they must be added by SQL commands or by database interface (e.g. Microsoft Access).
You can add new fields or update the existing ones by Paste col. button and also this function works only with SQL databases. The clipboard must contain data in text format in which the columns are separated by a single tab character and the rows by a single line feed. The first line must include the column/ field name and the number of rows must be the same of the number of molecules in the database. The data type and the field size are automatically detected and if the column is already present in the database, the pre-existing rows are updated with those of the clipboard.
The new field can be changed by clicking with the right mouse button on the field, showing the context menu:
| Menu item | Description |
| Rename | Rename the field/column. |
| Delete | Remove the field/column. |
WARNING:
You cannot edit the pre-defined of a SQL database.
The Reports tab allows to create a report of a whole database or a filtered subset (see below). There are some pre-defined reports that can be modified by the user in easy way (...\VEGA ZZ\Data\Databases\Reports directory).
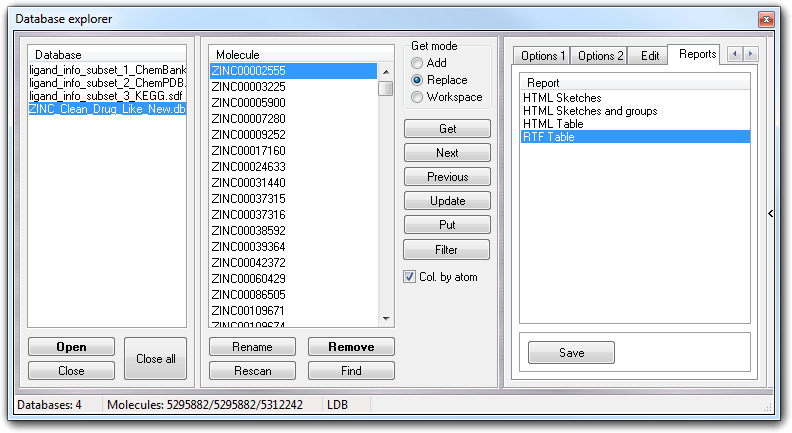
Click Save to save the report.
Databases containing millions of molecules are very hard to manage. For this reason, a function to filter/query a database was implemented. This function is more sophisticated than the Find feature (see the next section), because it allows to filter molecules on the basis of chemical-physical properties, composition and so on. It's available only for SQL databases (e.g. Access, SQLite, MySQL, ODBC, etc), because it requires some pre-calculated properties that aren't supported in other database formats such as SDF and Zip. To open Database SQL filter window, click Filer button:
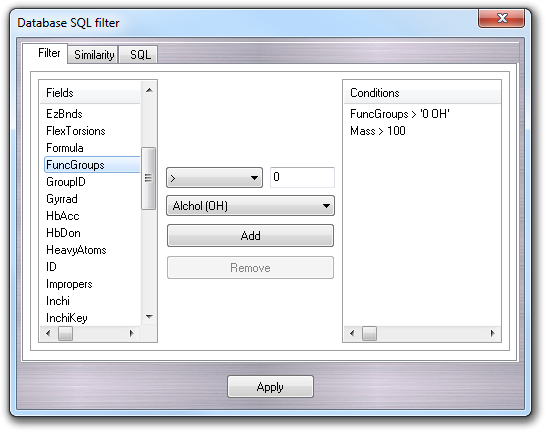
In Filter tab, it's possible to compose the query in easy way. In particular, you can select the property to filter clicking in the Fields column. There are some pre-calculated properties:
| Field | Type | Description | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Angles | I | Number of angles. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atoms | I | Number of atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bonds | I | Number of bonds. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Charge | F | Total charge. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ChiralAtms | I | Number of chiral atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Description | S | Molecule description. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Dipole | F | Dipole moment (Debye) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EzBonds | I | Number of bonds with E/Z geometry. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FlexTorsions | I | Number of flexible torsion angles. In other words, it's the number of rotable bonds. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Formula | S | Molecular formula. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FuncGroups | S | Functional group list. The string has the following
format (for internal use):NUM_1 GRP_1 NUM_2 GRP_2 ... NUM_N GRP_N where NUM is the number of functional groups of kind GRP. The functional groups are detected by the GROUPS.tem ATDL template (see the Data directory), as shown in the following table:
When you click this field, the list of the functional groups is automatically enabled to build the query in easy way. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GroupID | I | Group identification number. The molecules in a database can be grouped and each group has a unique identification number. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gyrrad | F | Gyration radius (Å). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HbAcc | I | Number of H-bond acceptor atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HbDon | I | Number of H-bond donor atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HeavyAtoms | I | Number of heavy atoms. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ID | I | Molecule identification number (primary key). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Impropers | I | Number of improper angles (out of plane). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Inchi | S | InChI string. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lipole | F | Lipole (lipophilicity moment). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mass | F | Molecular weight (Daltons). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molecules | I | Number of molecules. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Name | S | Name of the molecule. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ovality | F | Ovality (Å). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Psa | F | Polar surface area (Ų). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Rings | I | Number of rings in the molecule. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sas | F | Solvent accessible surface (Ų). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sav | F | Solvent accessible volume (ų). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sdiam | F | Surface diameter (Å). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Smiles | T | Smiles string. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Surface | F | Molecular surface (Ų). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Torsions | I | Number of torsion angles. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Vdiam | F | Volume diameter (Å). | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| VirtualLogP | F | Virtual logP. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Volume | F | Molecular volume (ų). |
where the Type column indicates field type: F = floating point number, I = integer number, S = character string.
More properties can be added by the user through SQL operations or by other software. They will be automatically available in the Fields column. After the selection of the property to filter, you can select the operator, put the value and click the Add button: the expression will be added to the Conditions column. There are some pre-defined operators:
| Operator | Description | ||||||||
| = | Equal. | ||||||||
| <> | Not equal. | ||||||||
| < | Less than. | ||||||||
| > | More than. | ||||||||
| <= | Less than or equal. | ||||||||
| >= | More than or equal. | ||||||||
| GLOB | This operator allows the pattern matching using the same syntax of
the Unix shell:
|
||||||||
| NOT GLOB | Not glob (see above). | ||||||||
| LIKE | Same function of GLOB, but it's possible to use the SQL
expressions:
|
||||||||
| NOT LIKE | Not like (see above). | ||||||||
| REGEXP | Same function of GLOB, but it's possible to use the regular expressions (RegExp). For more information, see http://www.regular-expressions.info/. | ||||||||
| NOT REGEXP | Not RegExp (see above). | ||||||||
| IS NULL | The field must be empty. | ||||||||
| NOT IS NULL | The field mustn't be empty. |
You can add more than one condition that are related each other by the AND logical operator. If you want edit a pre-entered condition, click it in the Conditions column and change the value and/or the operator. If you want remove a condition, click it and press the Remove button.
In the Similarity tab, it's possible to filter the molecules on the basis of their similarity with the structure in the current workspace. The implemented approach is based on the comparison of the fingerprint of the molecule in the current workspace with the those of the other ones in the database.
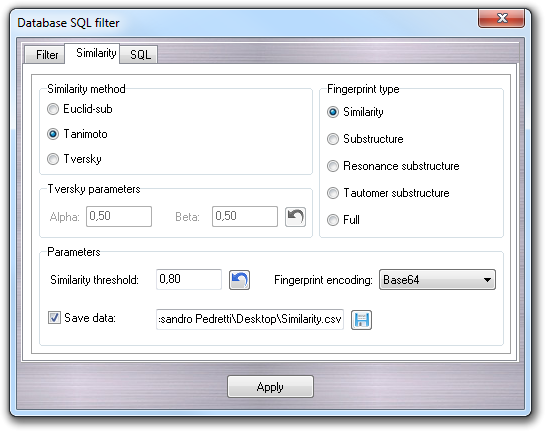
There are five different type of fingerprints that can be calculated (Similarity,
Substructure, Resonance substructure, Tautomer substructure
and Full) and three different metrics to compare the fingerprints, which
are calculated in the following way:
| Similarity method | Formula |
| Euclid-sub | c / a |
| Tanimoto | c / (a + b + b) |
| Tversky <alpha> <beta> | c / ((a - c) * alpha + (b - c) * beta) |
where a is the number of nonzero bits in the first fingerprint, b is the number of nonzero bits in the second fingerprint and c is the number of coincident bits in the two fingerprints. Alpha and beta are the weights of Tversky comparison.
In the Parameters box, you can set the Similarity threshold,
the fingerprint encoding method (Base64 or Hexadecimal) required
to save the fingerprints when you check Save data and put the output file
name for further analysis. The fingerprints, one for each molecule in the
database, are saved in CSV format.
In the SQL tab, it's possible to do more complex queries typing directly the SQL code:
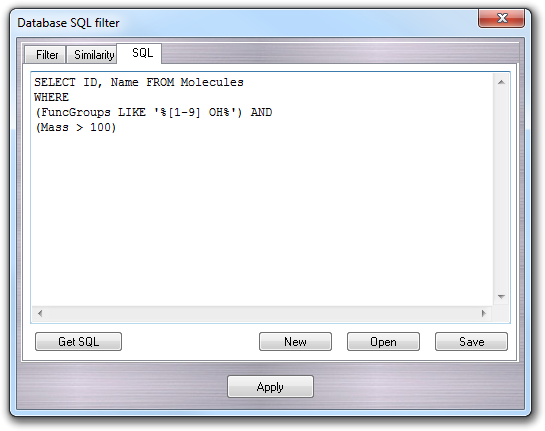
Finally, clicking the Apply button the query is performed and the molecules satisfying the user-defined conditions are shown in the Database explorer. To remove the filter, click the Rescan button.
A structure in the database can be updated selecting it in the molecule list
and clicking Update button. This structure will be replaced by the
molecule in the current workspace.
A molecule in the database can be renamed, selecting it and clicking Rename
button, but remember that the molecules included in a Zip file can't renamed.
In similar way, a molecule can be removed from the database selecting it and
clicking Remove button. Multiple selections are allowed.
Update button is useful to force the update of the molecule list when
the database was changed by another application.
To find molecules inside the active database, you can use the find function (Find
button, or Find item in the popup menu). The search is case-insensitive
and allows the use of wildcards (*, ? characters). Another method to find molecules
inside the database is available by the keyboard: typing a character on the
keyboard, the molecule with the name starting with that character is
automatically selected.
The status bar placed on the bottom of the window shows the total number of
opened databases, the total number of molecules, the number of molecules
selected by query, the number of molecules in the
active database and the database type indicator (SDB, LDB, see below).
Clicking with the right mouse button on the Database list or in the Molecule list, the context menu is shown:
| Database context menu | ||
| Item | Accelerator | Description |
| Open | Ctrl+O | Open a new database. |
| Close | Ctrl+C | Close the database. |
| Close all | - | Close all databases. |
| Synchronize | Ctrl+S | Synchronize the database (see the next section). |
| Molecule context menu | ||
| Item | Accelerator | Description |
| Get | Ctrl+G | Get the molecule. |
| Put | Ctrl+P | Put the molecule in the database. |
| Update | - | Update the molecule. |
| Rename | F2 | Rename the molecule. |
| Remove | - | Remove the selected molecules. |
| Filter | - | Show the filter dialog. This item appears checked if a filter was applied. |
| Find | Ctrl+F | Find a molecule in the list. |
| Rescan | - | Rescan the database to update the molecule list. |
| Save list | - | Save the molecule list to a file. |
| Copy list | - | Copy the molecule list in the clipboard. |
| Export to Excel | - | Export all molecule data to Microsoft Excel, excluding 3D structures. Miscrosoft Excel must be installed in your PC. |
| Export to file | - | Export all molecule data to a file, excluding 3D structures. You can choose different output formats: ARFF (Attribute-Relation File Format), ARFF without strings and CSV (Comma Separated Values). |
9.2.3 Small database and large database
VEGA ZZ can manage molecular databases in two modes:
Small database mode (SDB).
The small databases are updated in real time. If a molecule, is renamed,
removed or updated, the database is automatically rebuilt with the changes.
This operation requires a lot of time, so is applicable to small database
only.
Large database mode (LDB).
When you open a large database (size greater than 20 Mb), VEGA ZZ
scans it in order to build an index accelerating the next database accesses.
The obtained information is stored in a special IFF file called SDI (.sdi or
.dbi
extension). If you reopen the database, no rescan is required because VEGA
ZZ read the SDI file reducing dramatically the waiting time. When you
rename, remove or update a molecule, the database remains unchanged and the
modification are stored in the SDI file. In this way, the user obtains a
faster feedback. If the database must be used by another
software, it must be synchronized with the SDI file and to do it, you
must select Synchronize clicking with the right mouse button on the
database name.
The status bar highlight the operation mode showing the SDB or LDB labels. The LDB label could have a star (*), indicating that the selected database isn't synchronized. If the database is read-only, the Remove, Rename, Put, Synchronize and Update functions are disabled and in the status bar the RO label appears at the left of SDB/LDB word.
The database list is shared with the Add Fragment dialog. The fragment libraries are databases in zip format and so they appears in the database explorer. You can do all operations but you can't close them.
The database engine stores the file offsets in 64 bit integers breaking the limit of 2 Gb file size.
The maximum number of molecules for each database is 2^32 (4.294.967.296).
The maximum number of databases is 2^32 (4.294.967.296).
The maximum number of manageable molecules is 2^64 (18.446.744.073.709.551.616).
At this time, the SDF databases only are treated as SDB and LDB. The other database formats are always SDB.