12.6 FAME - Prediction of metabolism sites
12.6.1 Introduction
This plug-in interfaces VEGA ZZ to FAME 2 and FAME 3 programs in order to perform the prediction of sites of metabolism (SOMs) of a given molecule or a set of molecules. The input can be the molecule in the current workspace (in this case the SOMs are shown directly on the structure as transparent yellow spheres) or as file in SDF format (FAME 2 and 3) or as text file with the SMILES string and the name (only FAME 3). Both SD and text files can contain more than one molecule.
12.6.2 Requirements
Here are shown the hardware and software requirements.
12.6.2.2 Hardware requirements
FAME 2 and FAME 3 requires respectively at least 2 and 12 Gb of physical memory.
12.6.2.2 Software requirements
FAME plug-in requires VEGA ZZ 3.2.0 or
greater that must be pre-installed before the plug-in setup.
FAME 2 and FAME 3 programs require the
Java Runtime
Environment (JRE) and all dependencies are provided in the package. Since
FAME 3 requires more than 4 Gb of address space, to run it the 64 bit version of
JRE is needed.
12.6.3 The plug-in
12.6.3.1 Installation
The FAME plug-in is not included in the standard VEGA ZZ package and is provided as separated setup. In detail, there are two possible setup files:
Vega_ZZ_X.X.X.X_FAME_2.exe
and
Vega_ZZ_X.X.X.X_FAME_3.exe
where X.X.X.X is the VEGA ZZ version. The former is freely available and includes only FAME 2, while the latter includes both FAME 2 and 3 and is available on request. Before to run the plug-in setup, you must download and install VEGA ZZ.
12.6.3.2 Usage
To show the plug-in window, you must select
Calculation
![]() FAME in VEGA ZZ main menu. The plug-in recognizes automatically if FAME 2 or
3 or
both are installed, allowing you to select the program for a specific kind of
prediction (see Engine filed). In particular, FAME 2 recognizes only
the sites of oxidation in which the cytochrome P450 is involved, while FAME 3
predicts sites in which phase 1 and/or phase 2 metabolic reactions are involved.
FAME in VEGA ZZ main menu. The plug-in recognizes automatically if FAME 2 or
3 or
both are installed, allowing you to select the program for a specific kind of
prediction (see Engine filed). In particular, FAME 2 recognizes only
the sites of oxidation in which the cytochrome P450 is involved, while FAME 3
predicts sites in which phase 1 and/or phase 2 metabolic reactions are involved.
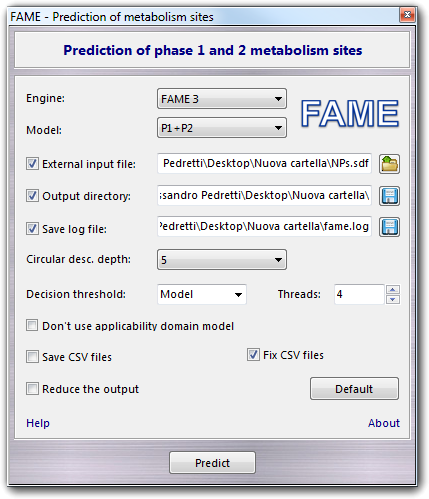
In the Model field, you can select the model for the prediction, according to the following table:
| FAME version | Model | Default | Description |
| 2 | circCDK_ATF_1 | Y | Offer the best trade-off between generalization and accuracy. It is based on the atom itself and its immediate neighbours (atoms at most one bond away). |
| 2 | circCDK_4 | N | One of the simpler model found to have comparable performance to the other ones. |
| 2 | circCDK_ATF_6 | N | Give the best average performance during the independent test set validation as performed in the FAME 2 paper or (circCDK_ATF_1 and circCDK_4). |
| 3 | P1+P2 | Y | Predict both phase 1 and phase 2 SOMs. |
| 3 | P1 | N | Predict phase 1 SOMs. |
| 3 | P2 | N | Predict phase 2 SOMs, |
By default, the plug-in performs the
prediction of SOMs of the molecule in the current VEGA ZZ workspace, but you can
use a file as input checking External input file and selecting a SDF or
SMILES file (SMILES files are supported only by FAME 3), which can contain one or more molecules. Checking Output
directory and selecting it, you can indicate where to save the output files,
otherwise they are stored in a temporary directory and deleted at end of
the calculation. Checking Save log file, you can specify the file to
which the FAME text output is saved. When you check Save CSV files, the used descriptors and
the prediction are saved in CSV format and you can decide to fix them according
the localization settings by checking Fix CSV files. When you have to
process a large number of molecules and you want to discard HTML files and the
data of non-SOM atoms, you can check Reduce the output. In this way, only
one file is saved (sites.csv).
To revert to the default settings, you can click the Default button and
to start the prediction, you must click the Predict button.
12.6.3.3 FAME 3 specific options
Here are explained the functions implemented only in FAME 3. In particular, the Circular desc. depth gadget allows you to choose between 2 and 5 as circular descriptor bond depth, which is the maximum number of layers to consider in atom type fingerprints and circular descriptors. The best results can be achieved with the default bond depth of 5, but in some cases the lower complexity model (2) could give better results especially if the FAMEscores are low. The Decision threshold, which must be in the range from 0 to 1, defines the decision threshold for the model. If you set it to Model, for the default value for the selected model is used. Checking Don't use the applicability domain model, the model to evaluate the applicability domain is switched off, the FAMEscore is not calculated and the prediction becomes faster. Finally, you can set the number of CPU cores/threads that are used for the calculation (Threads field). You can appreciate the FAME 3 parallelism only if you perform the prediction for more than one molecule.
12.6.4 FAME 3
This program attempts to predict sites of metabolism for the supplied chemical compounds. It is based on extra trees classifier trained for prediction of both phase I and phase II SOMs from the MetaQSAR database. It contains a combined phase I and phase II (P1+P2) model as well as separate phase I (P1) and phase II (P2) models. For more details on the FAME 3 method, see the FAME 3 [1] and MetaQSAR [2] publications:
Martin Šícho, Conrad Stork,
Angelica Mazzolari, Christina de Bruyn Kops, Alessandro Pedretti, Bernard
Testa, Giulio Vistoli, Daniel Svozil, and Johannes Kirchmair
"FAME 3: Predicting the Sites of Metabolism in Synthetic Compounds and
Natural Products for Phase 1 and Phase 2 Metabolic Enzymes"
Journal of Chemical Information and Modeling, Just Accepted Manuscript
DOI:
10.1021/acs.jcim.9b00376
Alessandro Pedretti, Angelica
Mazzolari, Giulio Vistoli, and Bernard Testa
"MetaQSAR: An Integrated Database Engine to Manage and Analyze Metabolic
Data"
Journal of Medicinal Chemistry, 2018, 61 (3), 1019-1030.
DOI:
10.1021/acs.jmedchem.7b01473
12.6.4.1 Installation
The installation of FAME 3 is not required because it is included in the plug-in setup. This section shows the installation of the command line version on other systems starting from a tar archive or the ...\VEGAZZ\ Fame 3 directory.
To install FAME 3, you must unpack the distribution archive:
tar -xzf fame3-${version}-bin.tar.gz ${YOUR_INSTALL_DIR}
alternatively, you can copy the "Fame 3" directory, which you can find in VEGA ZZ home folder to the your preferred installation directory.
On Linux and Macintosh platforms, running the program is easy since you can use the shell script provided in the installation directory:
cd ${YOUR_INSTALL_DIR}/fame3
./fame3
You can also add ${YOUR_INSTALL_DIR} to the $PATH environment variable to have universal access:
export PATH="$PATH:$YOUR_INSTALL_DIR"
To run FAME 3 on Linux and Macintosh, just type:
fame3 [OPTIONS]
On other platforms (e.g. Windows), you will have to run the java package explicitly:
java -Xmx16g -jar ${YOUR_INSTALL_DIR}\fame3.jar
Since the unpacked model takes several memory, the -Xms16g flag is necessary, overriding the default java options.
12.6.4.2 Command-line usage
If you run FAME 3 with the -h option, this help message is shown:
usage: fame3 [-h] [--version] [-m {P1+P2,P1,P2}] [-r PROCESSORS] [-d {2,5}]
[-s [SMILES [SMILES ...]]] [-n [NAMES [NAMES ...]]]
[-o OUTPUT_DIRECTORY] [-p] [-c] [-t DECISION_THRESHOLD] [-a]
[FILE [FILE ...]]
This is FAME 3 [1]. It is a collection of machine learning models to
predict sites of metabolism (SOMs) for supplied chemical compounds
(supplied as SMILES or in an SDF file).
FAME 3 includes a combined model ("P1+P2") for phase I and phase II SOMs
and also separate phase I and phase II models ("P1" and "P2"). It is based
on extra trees classifiers trained for regioselectivity prediction on data
from the MetaQSAR database [2].Feel free to take a look at the README.html
file for usage examples.
1. FAME 3: Predicting the Sites of Metabolism in Synthetic Compounds and
Natural Products for Phase 1 and Phase 2 Metabolic Enzymes
Martin èÝcho, Conrad Stork, Angelica Mazzolari, Christina de Bruyn Kops,
Alessandro Pedretti, Bernard Testa, Giulio Vistoli, Daniel Svozil, and
Johannes Kirchmair
Journal of Chemical Information and Modeling Just Accepted Manuscript
DOI: 10.1021/acs.jcim.9b00376
2. MetaQSAR: An Integrated Database Engine to Manage and Analyze Metabolic
Data
Alessandro Pedretti, Angelica Mazzolari, Giulio Vistoli, and Bernard Testa
Journal of Medicinal Chemistry 2018 61 (3), 1019-1030
DOI: 10.1021/acs.jmedchem.7b01473
positional arguments:
FILE One or more files with the compounds to predict.
FAME 3 currently supports SDF files and SMILES
files.In order for a file to be parsed as a
SMILES file, it needs to have the ".smi"file
extension. Files with a different extension will
be parsed as an SDF.The file can contain multiple
compounds.
All molecules should be neutral (with the
exception of tertiary ammonium) and have explicit
hydrogens added prior to modelling. However, if
there are missing hydrogens, the software will
try to add them automatically. Calculating
spatial coordinates of atoms is not necessary.The
compounds will be assigned a generic name if the
name cannot be determined from the file.
optional arguments:
-h, --help show this help message and exit
--version Show program version.
-m {P1+P2,P1,P2}, --model {P1+P2,P1,P2}
Model to use to generate predictions. Select
P1+P2 to predict both phase I and phase II SOMs.
Select P1 to predict phase I only. Select P2 to
predict phase II only. (default: P1+P2)
-r PROCESSORS, --processors PROCESSORS
Maximum number of CPUs the program should use.
Set to 0 to use all available CPUs. (default: 0)
-d {2,5}, --depth {2,5}
The circular descriptor bond depth. It is the
maximum number of layers to consider in atom type
fingerprints and circular descriptors. Optimal
results should be achieved with the default bond
depth of 5. However, in some cases the lower
complexity model could be more successful,
especially if FAMEscores are low. (default: 5)
-s [SMILES [SMILES ...]], --smiles [SMILES [SMILES ...]]
One or more SMILES strings of the compounds to
predict.
All molecules should be neutral (with the
exception of tertiary ammonium) and have explicit
hydrogens added prior to modelling. However, if
there are missing hydrogens, the software will
try to add them automatically. Calculating
spatial coordinates of atoms is not necessary.
-n [NAMES [NAMES ...]], --names [NAMES [NAMES ...]]
Use this parameter to provide names for compounds
submitted as SMILES strings.The number of
provided names needs to be the same as the number
of provided SMILES strings.
-o OUTPUT_DIRECTORY, --output-directory OUTPUT_DIRECTORY
Path to the output directory. If it doesn't
exist, it will be created. (default:
fame3_results)
-p, --depict-png Generates depictions of molecules with the
predicted sites highlighted as PNG files in
addition to the HTML output. (default: false)
-c, --output-csv Saves calculated descriptors and predictions to
CSV files. (default: false)
-t DECISION_THRESHOLD, --decision-threshold DECISION_THRESHOLD
Define the decision threshold for the model (0 to
1). Use "model" for the default model threshold.
(default: model)
-a, --no-app-domain Do not use the applicability domain model.
FAMEscore values will not be calculated, but the
predictions will be faster. (default: false)
12.6.5 FAME 2
This program attempts to predict sites of metabolism for supplied chemical compounds. It includes extra trees models for regioselectivity prediction of some cytochrome P450 isoforms. For more information on the method implemented in FAME 2, see the following publication:
Martin Šícho, Christina de Bruyn Kops, Conrad Stork, Daniel Svozil, Johannes Kirchmair
"FAME 2: Simple and Effective Machine Learning Model of Cytochrome P450 Regioselectivity"
Journal of Chemical Information and Modeling, 2017, 57 (8), 1832-1846.
DOI: 10.1021/acs.jcim.7b00250
12.6.5.1 Installation
As for FAME 3, the installation of FAME 2 is not required because it is included in the plug-in setup. This section shows the installation of the command line version on other systems starting from a tar archive or the ...\VEGAZZ\ Fame 2 directory.
To install FAME 2, you must unpack the distrubution archive:
tar -xzf fame3-${version}-bin.tar.gz ${YOUR_INSTALL_DIR}
alternatively, you can copy the "Fame 3" directory, which you can find in VEGA ZZ home folder to the your preferred installation directory.
On the Linux and Macintosh platforms, running the program is easy since you can use the shell script provided in the installation directory:
cd ${YOUR_INSTALL_DIR}/fame2
./fame2
You can also add ${YOUR_INSTALL_DIR} to the $PATH environment variable to have universal access:
export PATH="$PATH:$YOUR_INSTALL_DIR"
To run FAME 2 on Linux and Macintosh, just type:
fame2 [OPTIONS]
On other platforms (e.g. Windows), you will have to run the java package explicitly:
java -Xms1024m -jar ${YOUR_INSTALL_DIR}\fame2.jar
Since the unpacked model takes quite a bit of memory, the -Xms1024m flag is necessary, overriding the default java options.
12.6.5.2 Command-line usage
If you run FAME 2 with the -h option, this help message is shown:
usage: fame2 [-h] [--version] [-m {circCDK_ATF_1,circCDK_4,circCDK_ATF_6}]
[-s [SMILES [SMILES ...]]] [-o OUTPUT_DIRECTORY] [-p] [-c]
[FILE [FILE ...]]
This is fame2. It attempts to predict sites of metabolism for supplied
chemical compounds. It includes extra trees models for regioselectivity
prediction of some cytochrome P450 isoforms.
positional arguments:
FILE One or more SDF files with compounds to predict.
One SDF can contain multiple compounds.
All molecules should be neutral and have explicit
hydrogens added prior to modelling. If there are
still missing hydrogens, the software will try to
add them automatically.Calculating spatial
coordinates of atoms is not necessary.
optional arguments:
-h, --help show this help message and exit
--version Show program version.
-m {circCDK_ATF_1,circCDK_4,circCDK_ATF_6}, --model {circCDK_ATF_1,circCDK_4,circCDK_ATF_6}
Model to use to generate predictions.
Either the model with the best average
performance ('circCDK_ATF_6') during the
independent test set validation as performed in
the original paper or one of the simpler models
that were found to have comparable performance
('circCDK_ATF_1' and 'circCDK_4'). The
'circCDK_ATF_1' model is selected by default as
it is expected to offer the best trade-off
between generalization and accuracy.
The number after the model code indicates how
wide the encodedenvironment of an atom is. For
example, the default 'circCDK_ATF_1' is a model
based on the atom itself and its immediate
neighbors (atoms at most one bond away).
(default: circCDK_ATF_1)
-s [SMILES [SMILES ...]], --smiles [SMILES [SMILES ...]]
One or more SMILES strings of compounds to
predict.
All molecules should be neutral and have explicit
hydrogens added prior to modelling. If there are
still missing hydrogens, the software will try to
add them automatically.
-o OUTPUT_DIRECTORY, --output-directory OUTPUT_DIRECTORY
The path to the output directory. If it doesn't
exist, it will be created. (default: fame_results)
-p, --depict-png Generates depictions of molecules with the
predicted sites highlighted as PNG files in
addition to the HTML output. (default: false)
-c, --output-csv Saves calculated descriptors and predictions to
CSV files. (default: false)
12.6.5.3 Examples
If you want to perform the prediction for a single molecule, which must be in SDF format, you must type in the command prompt:
fame2 -o test_predictions tamoxifen.sdf
where test_predictions is the directory in which the output files are saved and tamoxifen.sdf is the input file including the structure of the molecule that can be 2D or 3D. You must remember that the molecule must have explicit hydrogens and must be in neutral form whit the exception of the quaternary nitrogens.
The program also accepts also SMILES strings as input:
fame2 -o "test_predictions" -s CCO c1ccccc1C
This creates the test_predictions folder in the current directory which contains the output files for each analyzed compound.
12.6.6 Copyright and disclaimers
The FAME software is based on a number of
third-party dependencies that are listed in the NOTICE
document, which also includes their licensing information and links to web sites
where original copies of the software can be obtained. The source code of the
third-party libraries was not modified with the important exceptions of the
SMARTCyp software and some classes from the WEKA machine learning
library (version 3.8).
The SMARTCyp code was slightly adapted in order to work well with
the FAME 3 software and the changes are tracked in the
publicly available source code repository.
From the WEKA library, only the LinearNNSearch class was modified for
thread safety.
The SMARTCyp code was obtained through the SMARTCyp web site
mentioned in the
original publication.
The source files to be modified from the WEKA library were obtained from
GitHub.
FAME 2 and FAME 3
are pieces of software developed in 2017-2021
by Martin Šícho & Johannes Kirchmair
All rights reserved.
Martin Šícho
Z-OPENSCREEN: National Infrastructure for Chemical Biology
Laboratory of Informatics and Chemistry,
Faculty of Chemical Technology, University of Chemistry and Technology Prague
166 28 Prague 6, Czech Republic
E-mail: martin.sicho@vscht.cz
Johannes Kirchmair
Universität Hamburg,
Faculty of Mathematics, Informatics and Natural Sciences
Department of Computer Science, Center for Bioinformatics
Hamburg, 20146, Germany
E-Mail:
kirchmair@zbh.uni-hamburg.de
Click here to read the complete FAME license.
FAME plug-in
for VEGA ZZ
is a software developed in 2018-2021
by Alessandro Pedretti & Giulio Vistoli
All rights reserved.
Alessandro Pedretti
Dipartimento di Scienze Farmaceutiche
Facoltà di Scienze del Farmaco
Università degli Studi di Milano
Via Luigi Mangiagalli, 25
I-20133 Milano - Italy
E-Mail: info@vegazz.net
Click here to read the complete VEGA ZZ license.