GriDock
The virtual screening front-end for AutoDock 4
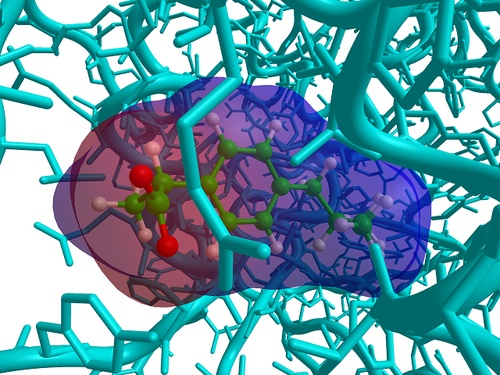
GriDock was designed to perform the molecular dockings of a large number of ligands stored in a single database (SDF or Zip format) in the lowest possible time. It take the full advantage of all local and remote CPUs through the MPICH2 technology, balancing the computational load between processors/grid nodes. The docking procedure consists in some steps:
- Extraction of the molecule from the database (VEGA command line).
- Addition of the hydrogens if they are missing (VEGA command line).
- Conversion from 2D to 3D (if required) provided by AMMP VE.
- Optional structure filter based on the chemical/physical properties calculated by VEGA.
- Atomic charges calculation, potential attribution (AMBER), search of the flexible torsion angles and conversion to PDBQT format (VEGA command line).
- Molecular docking (AutoDock 4).
- Selection of the best scored complex and ranking in the result list.
- Storing of the AutoDock output in a compressed Zip archive for a successive analysis (e.g. with VEGA ZZ).
Main features:
- No database pre-processing needed. The ligands can be stored in 2D or 3D with or without hydrogens.
- No external scripts. GriDock is a monolithic softtware written in C++ and it's based on the VegaLib runtime.
- Easy to use even if it's a command line-based software. It requires two parameters only to run: the receptor file name and the ligand database.
- Windows executable for both thread and MPICH2.
- Linux 32 and 64 bit executables for both pthread and MPICH2.
Software requirements:
- AutoDock 4 and AutoGrid 4.
- VEGA 2.3.2 command line version.
- AMMP VE.
System requirements:
- Multi core/CPU system or grid system.
- Windows (32 or 64 bit) or Linux operating system (32 or 64 bit).
- MPICH2 if you want use the MPI version on a grid.
GriDock is included in VEGA ZZ for Windows and VEGA 3.0 for Linux x86-x64-ARM packages.
>>> Click here to download it <<<
|