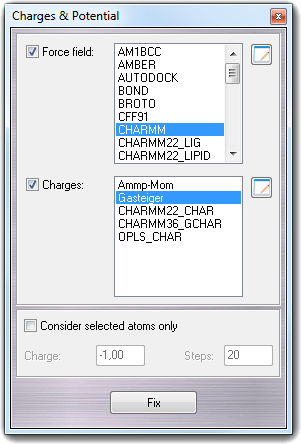
8.1 Charges & potential
By this dialog, it's possible to assign atom types and atomic charges. To open it, select Charge & Pot. in the Calculate menu.
Check and select the Force field type if you want fix the atom types for molecular mechanics calculations. To assign the atomic charges, you must check Charges and select the method. Some methods are available to assign the atomic charges:
| Method | Description |
| Ammp-Mom | It's based on the bond polarization and it's performed by the AMMP package. When you select this method, you must specify the total charge (Charge field) and the number of steps (Steps field) for the charge optimization. This method isn't recommended for large molecules because it's very expensive in term of time. |
| Formal | Assign formal charges only. |
| Gasteiger | It's the Gasteiger-Marsili method, it's universal and it uses typed atomic charges that are smoothed on polar atoms. |
| CHARMM22_CHAR | It uses the pre-computed atomic charges included in the CHARMM 22 force field. It's less flexible because it uses a fragment/residue database containing the pre-computed charges. If a residue/fragment is not included in the database, the atomic charges can't be assigned. |
| OPLS_CHAR | It uses the pre-computed atomic charges included in the OPLS force field and it has got the same limits of the previous one. |
If you want to fix
atom types and charges to selected atoms only, you must check Consider
selected atoms only. Press Fix to calculate the atom types and/or
the charges. Clicking the button
![]() , it's possible to modify the parameters of the selected
template/data file through the MiniEd:
, it's possible to modify the parameters of the selected
template/data file through the MiniEd:
Please remember to save the changes in order to activate the modifications. The Force Field combo box is populated scanning the ...\VEGA\Data directory every time that the dialog is opened. If a new template is added (click here for more information) and VEGA ZZ is running, you must close and reopen the dialog to refresh the list.