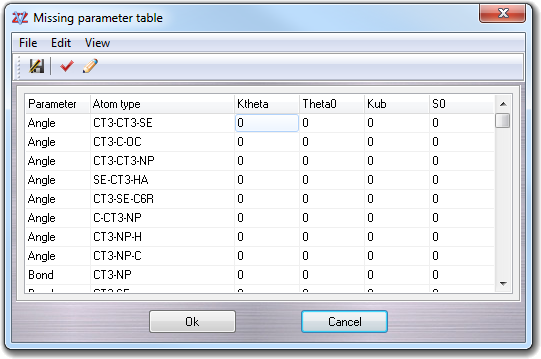
6.13 Missing parameter table
VEGA ZZ shows this table when a molecular mechanics calculation is starting (e.g. AMMP minimization) or a force field parameter check is requested (e.g. saving a molecule) and one or more parameters are missing in the current parameter file:
Each missing parameter occupies a line in the table that is subdivided in six columns: the first two columns have always the same meaning (Parameter and Atom type), and the other columns change the meaning on the basis of the parameter type (e.g. angle, bond, torsion, etc.) as reported in the following table:
| Parameter type | Column 3 | Column 4 | Column 5 | Column 6 |
| Angle | Ktheta Force constant, kcal/mol/rad2 |
Theta0 Angle value, degrees |
Kub Urey-Bradley force constant, kcal/mol/Å2 (not used by AMMP) |
S0 1-3 distance, Å (not used by AMMP) |
| Bond | Kb Force constant, kcal/mol/Å2 |
b0 Bond length, Å |
- | - |
| Torsion (dihedral) | Kchi Force constant, kcal/mol |
n Multiplicity |
delta Torsion offset, degrees |
- |
| Hybrid (improper, out of plane) | Kpsi Force constant, kcal/mol/rad2 |
n Multiplicity |
psi0 Hybrid offset, degrees |
- |
| Nbond (non-bond) | eps epsilon, kcal/mol |
Rmin/2 half distance, Å |
eps 1-4 epsilon 1-4, kcal/mol (not used by AMMP) |
Rmin/2 1-4 half distance 1-4, Å (not used by AMMP) |
Moving the cursor and clicking over the cells, the missing parameter is highlighted in the structure:
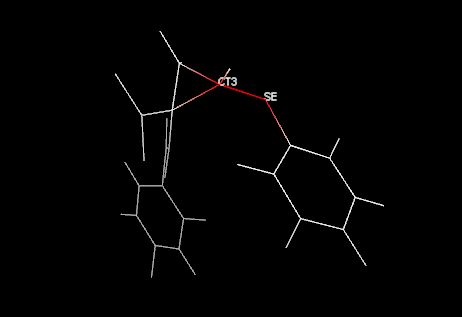
This is a picture example resulting by the
click on the CT3-SE bond missing parameter cell. This behaviour can be changed
un/checking the Color atoms, Labels and Wireframe items in
the View menu: the first one allows to color the highlighted structure in
red and the other atoms in white, the second one shows the atom type labels and
the third one change the view to wireframe.
You can type the parameters selecting each cell, but if you don't have any
idea about those values, the context menu can suggest them for you, clicking
the cell with the right mouse button.
Another way to fix the parameters is the use of the automatic assignment. This
function is accessible selecting Edit
![]() Auto assign in the menu bar. The
method is a little bit dirty, but it's a good starting point to refine the
missing parameters.
Auto assign in the menu bar. The
method is a little bit dirty, but it's a good starting point to refine the
missing parameters.
The new parameters can be saved in CHARMM format (*.inp) selecting File
![]() Save As ... menu item. In this way, you can merge the parameters with the
user parameter file in the VEGA ZZ\Data\Parameters directory, in order to
avoid to repeat the same operation for similar molecules (see
InpMerge utility). You can use them with NAMD also:
Save As ... menu item. In this way, you can merge the parameters with the
user parameter file in the VEGA ZZ\Data\Parameters directory, in order to
avoid to repeat the same operation for similar molecules (see
InpMerge utility). You can use them with NAMD also:
# This is a simple NAMD 2 configuration file # to perform an energy minimization numsteps 8000 minimization on dielectric 1.0
coordinates molecule.pdb outputname molecule outputEnergies 1000 binaryoutput no DCDFreq 1000 restartFreq 1000
structure molecule.psf paraTypeCharmm on parameters par_all22_prot.inp parameters molecule.inp exclude scaled1-4 1-4scaling 1.0 switching on switchdist 8.0 cutoff 12.0 pairlistdist 13.5 margin 0.0 stepspercycle 20
The red line indicates the file with the parameters not included in the standard par_all22_prot.inp file and previously generated saving the missing parameter table. The PDB and the PSF files (blue) can be generated by VEGA ZZ using the save molecule function.
Clicking Ok button or selecting
File
![]() Continue menu item, the procedure invoking the missing
parameter table continues using the parameters edited by the user. Clicking Cancel button or selecting
File
Continue menu item, the procedure invoking the missing
parameter table continues using the parameters edited by the user. Clicking Cancel button or selecting
File
![]() Cancel menu item, the procedure
is aborted.
Cancel menu item, the procedure
is aborted.