8.2
AMMP calculation
AMMP is a modern full-featured molecular mechanics, dynamics
and modelling program. It can manipulate both small molecules and macromolecules
including proteins, nucleic acids and other polymers. In addition to standard
features, like numerically stable molecular dynamics, fast multipole method for
including all atoms in the calculation of long range potentials and robust
structural optimizers, it has a flexible choice of potentials and a simple yet
powerful ability to manipulate molecules and analyze individual energy terms.
One major advantage over many other programs is that it is easy to introduce
non-standard polymer linkages, unusual ligands or non-standard residues. Adding
missing hydrogen atoms and completing partial structures, which are difficult
for many programs, are straightforward in AMMP. For more information, see the
AMMP manual.
Selecting an item in the Calculate
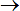 Ammp
menu, the AMMP dialog windows is shown. At this time, it's possible to
do energy minimization and conformational search only, but other calculation modes are
accessible trough the AMMP direct commands (see the
AMMP manual).
Ammp
menu, the AMMP dialog windows is shown. At this time, it's possible to
do energy minimization and conformational search only, but other calculation modes are
accessible trough the AMMP direct commands (see the
AMMP manual).
Please remember that before to perform an AMMP calculation, the atom charges
must be assigned (for more information, click here).
The atom types don't need to be assigned, because they are automatically recognized
every time that a calculation starts. If you
find problems in the automatic assignment, you can proceed to fix them assigning
the atom types using the Calculate
Charge & Pot. menu item (SP4
force field) or the manual function (Edit
Change
Atom/Residue/Chain).
WARNING:
the SP4 force field is atom-oriented: it means that the force constants are
computed starting from the atoms parameters and they aren't in angle, bond,
torsion and improper tables. To compute that constants, the bond order is
required. Optimizations of molecules with wrong bond types (single, partial
double, double and triple) could carry out to bad structures. If you need to
fix the bond types, select Edit
Change
Bonds in the menu bar,
choose Find the bond types and finally click the Apply button (for
more details, click here).
The bond types are automatically checked before starting the minimization. If a
problem is found, a warning dialog window is shown by which it's possible to
ignore the problem or to abort the procedure highlighting the atoms with the
possible wrong bond order.
8.2.1 Energy minimization
To perform an energy minimization,
select Minimization in Calculate
Ammp menu.
In the Minimization tab,
you can change the main minimization parameters and the minimization
algorithm: Single point,
Steepest descent,
Trust,
Conjugate gradients,
Quasi-Newton,
Truncated Newton,
Genetic algorithm,
Polytope simplex and
Rigid-body. The Graphic
update field sets the number of iterations after which the VEGA ZZ
3D view is refreshed (nupdat
variable).
When you start the minimization clicking the Run button, the
calculation steps and the error messages are shown in VEGA ZZ
console. If you want to stop the interactive run, you must click with the
right mouse button on the workspace area and select Stop calculation.
In the same way, it's possible to shutdown the system when the calculation is
finished, checking Power off the system.
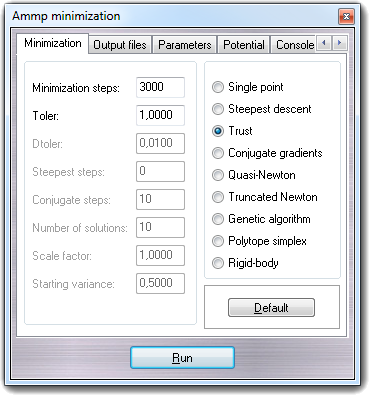
Is it possible to save the Trajectory, the Output messages
generated by AMMP and shown in the console, the Energy for each
trajectory frame and the Velocities (this option isn't yet
available), enabling the each field clicking the checkbox. To change the
file names and the file formats, click the disk button
 .
.
If you are changing the trajectory file name, the Save trajectory file
requester is shown, in which you can select the trajectory file format
(for more information click here). To copy the
file name to all fields, click the small button
 at the left of the disk
button.
To revert to the default parameters, click the Default button.
at the left of the disk
button.
To revert to the default parameters, click the Default button.
WARNING:
the fields of output files are automatically filled using the file path of
the last loaded molecule and the name of the current workspace. If you change
the current workspace, the file names are updated and manual changes are lost.
8.2.2
AMMP conformational search
To perform a conformational search,
select Conformational search in Calculate
Ammp
menu.
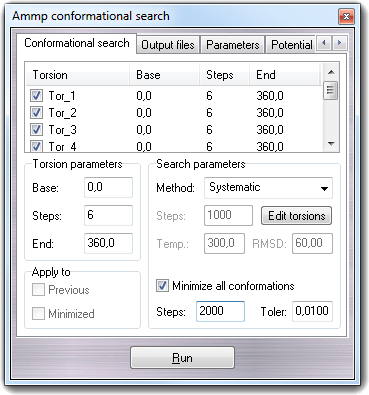
In Search parameters, you can select the search method (Method
combo-box): Systematic,
Random and
Boltzmann jump.
As first step, you must select the torsion (dihedral) angles that will be considered
during the conformational search. To do it, press the Edit torsions
button: the Selection tool dialog window will
be shown. You can also load the selection by drag & drop of a file or by
context menu (Open menu item). By this tool, you can choose all or
flexible or user defined torsions and the list is automatically updated
in the conformational search windows also. For each
torsion, the name (Torsion), the starting value in degrees (Base),
the number of rotation steps for the systematic search (Steps)
and the range in degrees in which the random rotations are computed (Window)
or ending rotation value (End) when you perform the systematic search.
The checkbox at the beginning of each line allows to consider or not the torsion
in the calculation. With the context menu, you can un/check all torsions
(Check all, Uncheck all), un/check the
highlighted/selected torsions (Check selected, Uncheck
selected) and un/select all torsions (Select all, Unselect
all). Highlighting one or more torsions in the
list, you can change the parameters (Base, Steps and Window)
in the Torsion parameters box. Not all parameter fields are enabled at the same time
and that's related to the type of the search that you selected (see Method
combo-box).
Checking Minimize all conformations, a
conjugate gradients minimization is
performed for each generated conformation. You can set the number of
minimization steps (Steps) and toler value (Toler). Generally it's
strongly recommended to enable this option.
Choosing the random search method, the Steps field in the Search
parameters box is enabled, in which you can specify the number of random
conformations that will be generated. In the Apply to box, you can decide
to apply the random rotation to the starting conformation (none checked), to the
previous generated conformation (Previous checked) and to the previous
minimized conformation (Previous and Minimized checked). If
Minimize all conformations is not checked, the meaning of this last option
is the same of Previous checked only.
Choosing the Boltzmann jump method, the Steps, Temp. and RMSD
fields are enabled: Steps allows to specify the number of conformations
that will be generated, Temp. is the temperature in Kelvin and RMSD
is the torsion root mean square difference (in degrees) used in the Boltzmann
jump perturbation phase to generate a significant different conformation
compared to the previous one.
Clicking the Run button, the conformational search begins and at the end
the lowest energy structure is kept in the workspace.
Some operations, shown in the next table, can be done by the context menu
that can be displayed by clicking with the right mouse button on the torsion
list:
| Item |
Subitem |
SubSubItem |
Description |
| Open
|
- |
- |
Open a torsion selection (for more information, see the
Selection tool). |
| Save as ... |
- |
- |
Save the torsions to a file. |
| Fill |
Base & end |
± 5° |
Systematic: complete automatically Base and End
fields of each highlighted torsion with the range obtained from the
current torsion angle adding and subtracting the selected value.
Random and Boltzmann jump:
complete automatically Base and Window fields of each
highlighted torsion respectively with the current torsion angle and the
double of selected value.
|
| ± 10° |
| ± 20° |
| ± 30° |
| ± 45° |
| ± 60° |
| ± 90° |
| ± 120° |
| ± 80° |
| Base with torsion value |
- |
Complete automatically the Base field of each highlighted torsion
with the current torsion value. |
| Current values |
- |
Set Base, Steps and Window/End fields of each highlighted
torsion with the values in the Torsion parameters box. |
| Default |
- |
Set Base, Steps and Window/End fields of each highlighted
torsion with the default values (respectively 0.00, 6, 360.00). |
| Check all |
- |
- |
Check/enable all torsions. |
| Check selected |
- |
- |
Check/enable the selected torsions. |
| Uncheck all |
- |
- |
Uncheck/disable all torsions. |
| Uncheck selected |
- |
- |
Uncheck/disable the selected torsions. |
| Select all |
- |
- |
Select all torsions. |
| Unselect all |
- |
- |
Unselect all torsions. |
WARNING:
it's strongly recommended
to set the Graphic update to 1, otherwise not all conformations are
saved in the output file, but only one every N conformations, where N
is the graphic update value.
8.2.2.1 Conformational search example
Imagine to perform a conformational search
of a small molecule using the Boltzmann jump method:
-
Open or build the molecule. If you
want use the SP4 force field (recommended), check if the bond types (bond
order) are correctly assigned. If it's not true, select Edit
Change
Bonds in the main menu, choose Find the
bond types, click Apply.
-
Fix the atom types (optional) and the
charges (Calculate
Charge & Pot.).
-
If the starting geometry isn't
minimized, before the conformational search, perform a full minimization. To
do it, open the minimization dialog window (Calculate
Ammp
Minimization), uncheck the outputs (Trajectory, Output and
Energy), select Conjugate gradients, 3000 steps, 0.01 toler, 0
steepest steps and finally click the Run button.
-
Open the conformational search dialog
window (Calculate
Ammp
Conformational search).
-
Add all flexible torsions clicking the
Edit torsion buttons. For more information about the selection tool
click here.
-
In the Search parameters box,
set Method = Boltzmann jump, Steps = 1000 (default value), Temp. = 1000 and
RMSD = 60 (default value).
-
Check Minimize all conformations,
Steps = 50, Toler = 0.01 (default value).
-
Check Trajectory, Output
and Energy outputs. Change the default file name and or the
trajectory format if it's required by you.
-
Click the Run button.
-
At the end of the calculation, three
files will be obtained: a trajectory file (the
trajectory analysis tool can open it), an output file containing all
messages printed in the console by AMMP and an energy file in CSV format
(the first value is the conformation number and the second one is the
energy).
-
The resulting conformations can be
finally clustered (see Trajectory analysis).
8.2.3 AMMP console
In the Console tab, it's possible to control AMMP sending direct
commands.
This function is useful to perform operations not implemented in the
graphic user interface or to get/set the system variables. The output is always
redirected to VEGA ZZ console.
The commands must be typed in the bottom box and confirmed clicking
Send or pressing the return key. The top box is the command history
containing the latest typed commands. They can be repeated double clicking the
line.
8.2.4 Calculation parameters
The Parameters tab allows
to change the Dielectric constant (dielect
variable), the Long range cutoff (cutoff
variable), the Short range cutoff (mxcut
variable), the Update full electrostatic threshold (mxdq
variable), the Lambda value for homotopic force field terms (lambda
variable) and Random number seed (seed
variable). This value is used to initialize the pseudo-random number
generator.
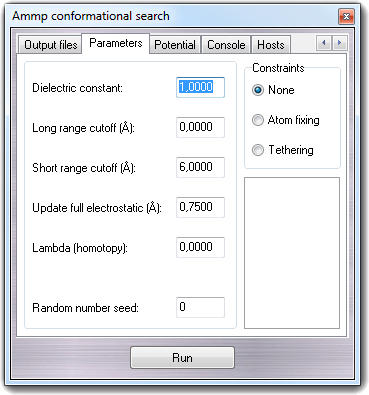
In Constraints box, it's possible to enable the use of the
constraints defined by the Atom constraints
function: None makes all atoms free without constraints,
Atom fixing keep the atoms totally fixed (see
ACTIVE and
INACTIVE commands) and
Tethering allows little movements around the starting position of
the atoms depending on force constants (see
TETHER command).
To revert to default parameters, click the Default button.
8.2.5 Potential terms
In Potential tab, it's possible to change the potential terms used
for the energy evaluation.
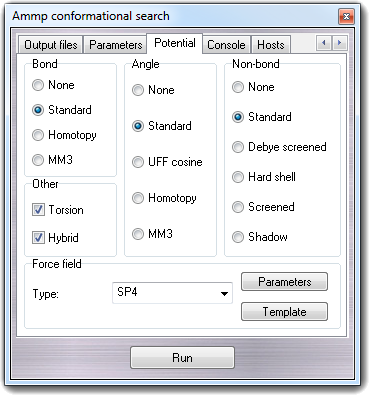
For more details, see the AMMP's USE
command. In the Force field box, you can change the force field
type used by AMMP: SP4 (the standard AMMP force field) and CHARMM 22. The Template
button allows to change the ATDL template used for the atom type
recognition, the Parameters button allows to change the potential parameters.
To revert to default parameters, click the Default button.
8.2.5.1 Non-SP4 force fields
The
parameter files of these force fields must be in the standard CHARMM/NAMD
format. They must placed in the VEGA ZZ\Data and in the VEGA ZZ\Data\Parameters
directories and they must have the .inp extension. For the automatic atom
type assignment, for each .inp file must exist a .tem file containing the ATDL
tremplate (e.g. CHARMM22_PROT.inp and CHARMM22_PROT.tem). The parameter files
interpreter included in the HyperDrive library has the pre-processor feature to
include more files:
*
* CHARMM 22 parameters file for VEGA ZZ
*
INCLUDE "Parameters/par_all22_lipid.inp"
INCLUDE "Parameters/par_all22_na.inp"
INCLUDE "Parameters/par_all22_prot.inp"
INCLUDE "Parameters/par_all22_vega.inp"
INCLUDE "Parameters/par_all22_user.inp"
WARNING:
no error message is shown
if the included file doesn't exist. In the VEGA ZZ\Data\Parameters directory,
custom parameter files can be placed:
*_vega.inp are created by the VEGA team to expand the standard parameters and *_user.inp can be generated by users when the
parameters are missing in the standard files.
When a non-SP4 force field is selected, is it possible
that one or more parameters are missing and so the
missing parameter table is shown in order to allow to the user to add the
missing parameter.
8.2.6 Hosts
AMMP can be executed on local and
remote hosts according to the VEGA ZZ calculation host concept (for more
information, click here).
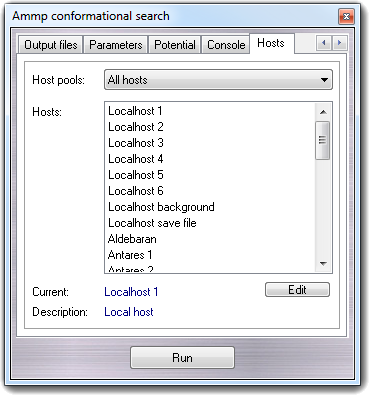
If you select a
local host, the calculation is executed using the local hardware,
otherwise is executed on a remote host. Host pools allows to
filter the host list and the default pools are: All hosts,
Local hosts and Remote hosts. Other polls can be defined by
the user for massive parallel calculations not yet implemented.
Selecting Localhost the calculation is immediately
executed in interactive mode (if you close VEGA ZZ, the job is stopped);
selecting Localhost background, the calculation is started in background
and it can't be controlled by VEGA ZZ (if you close VEGA ZZ, the job continues
without stopping itself); selecting Localhost save file, the calculation
is not started and the input file is created to run the job later.
Please note the duplicated hosts: they are child hosts and they
are assigned one for each physical or virtual CPU (for more information,
click here).
The Edit button allows to open the host
configuration window.