8.3 NAMD calculation
NAMD is a parallel molecular dynamics code designed for high-performance
simulation of large biomolecular systems. NAMD scales to hundreds of processors
on high-end parallel platforms, as well as tens of processors on low-cost
commodity clusters, and also runs on individual desktop and laptop computers.
NAMD works with AMBER and CHARMM potential functions, parameters, and file
formats (from J. of Compt. Chem. Vol. 26 (16), 1781-1802, 2005).
VEGA ZZ integrates in its environment the powerful capabilities of NAMD to
perform molecular dynamics and energy minimizations in interactive mode. The
graphic user interface allows to access to NAMD configuration in easy way
and the 3D view permits to show the structural changes during the simulation
by IMD interface (Interactive Molecular Dynamics). Through the
included pre-settings, the users with a very basic knowledge of the MD theory
can perform simulations and analyze the results as explained in the
Trajectory analysis section.
Nevertheless, it's strongly recommended to consult the
NAMD User Guide
available at Theoretical and
Computational Biophysics Group to understand better the MD concepts and the
meaning of the parameter fields accessible by the graphic interface.
Selecting Calculate
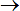 NAMD
in the main menu or clicking the icon in the left toolbar, NAMD dialog window is
shown.
NAMD
in the main menu or clicking the icon in the left toolbar, NAMD dialog window is
shown.
Some information included in this manual section, are from NAMD User Guide.
8.3.1 Before to run a NAMD simulation
Before to start a NAMD calculation, the molecule that you want to undergo to the
simulation must be prepared:
- it must be completed (check hydrogens and connectivity). The bond
order isn't used by NAMD and so it can remain unassigned (remember
that AMMP has a completely different behaviour: it requires the bond order to calculate the
force constants);
- the atom charges must be assigned/calculated (see
Charges and potential and
Mopac for small molecules);
- all atoms must have the correct atom type that can be assigned manually
(Charges and potential) or automatically by NAMD interface. The automatic assignment is enabled when the interface finds
atom type unassigned or included in a force field that differs from that you
selected in the dialog window;
- is not possible to perform simulations even if one atom has an
unassigned potential. They are highlighted when you assign the atom types
with the Charges and potential function with the question mark label
(?) or showing the atom types labels (View
Label atom
Type in
main menu).
WARNING:
not all atom types are supported by NAMD: you can use CHARMM22_LIG
(includes parameters for small molecules, proteins and nucleic acids),
CHARMM22_LIPID (for lipids only), CHARMM22_NA (for nucleic acids
only), CHARMM22_PROT (for proteins only) and OPLS. The CHARMM
force field can be used only if you have the Accelrys' parm.prm file in
the ...\VEGA ZZ\Data\Parameters directory that can't be included in the
package for copyright reasons.
No special files are required to run the simulation (e.g. PDB and PSF files)
because all needed ones are created by the interface.
8.3.2 NAMD interface basics
The interface is designed to remember the input fields/commands used in the
standard NAMD input files: moving the mouse pointer over a field label a hint
with the name of the NAMD command is shown.
Context menus are accessible making faster the page navigation:
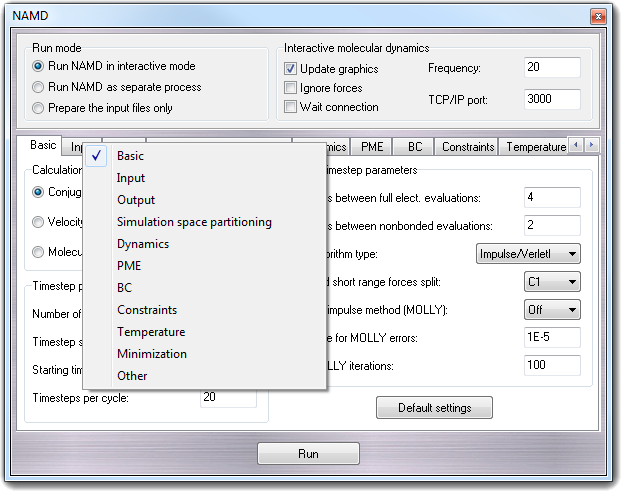
Clicking the right mouse button outside the page bars, the settings menu
is shown:
This menu allows to load (Load settings) or save
(Save settings) the settings in standard NAMD configuration file format. Checking
the Ignore file names, the commands having a file name as argument
are ignored (this is useful to keep the file names in the dialog window).
Selecting Default settings, all parameters are reverted to the default
values.
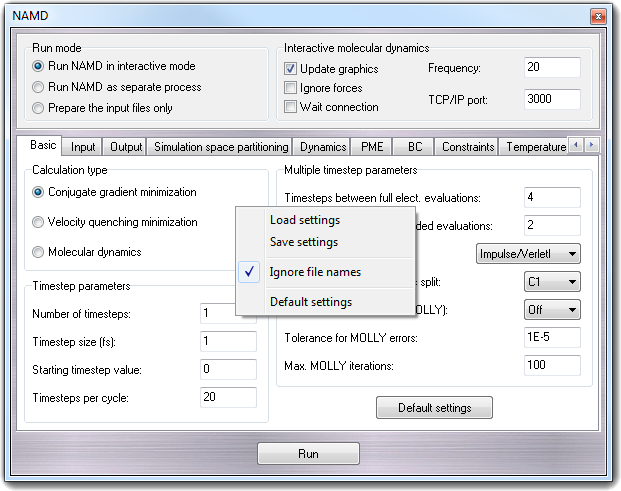
8.3.3 Run modes and Interactive
Molecular Dynamics (IMD)
The box at the top of NAMD dialog window allows to change the run mode and
the IMD parameters:
The Run mode group set how NAMD is started when the you
click Run button:
- Run NAMD in interactive mode.
It's the default operation mode. VEGA ZZ creates a process attached to NAMD
and if the IMD is enabled, the 3D graphic is updated during the simulation.
When VEGA ZZ is closed, the NAMD process is killed and the simulation
aborts.
- Run NAMD as separated process.
VEGA ZZ creates a detached process for NAMD. No graphic update is possible,
but the calculation isn't killed if VEGA ZZ is closed.
- Prepare the input files only.
Choosing this option, the NAMD input files are created without to start the
calculation. That's useful to prepare one or more calculations for batch
run. In the directory where the files are placed, a file with the
calculation prefix and the .cmd extension as name is saved and
executing it, you can start the calculation.
In Interactive molecular dynamics box, checking Update graphics
(correspondent to the IMDon NAMD command), you can enable the
visualization of the structure changes in the 3D environment during the
simulation. Frequency (IMDfreq) sets the graphic update frequency
(e.g. 20 means that the graphic is updated every 20 simulation steps), TCP/IP
port (IMDport) sets the communication port number between NAMD and VEGA ZZ. If
the port is already in use by another application, its value is automatically
increased until a free port is found. Checking Ignore forces (IMDignore), NAMD ignores the forces sent by the client application (VMD) for the steered
molecular dynamics and checking Wait connection (IMDwait), NAMD
stops itself in initialization phase until the connection with the
client application is completed (VMD or VEGA ZZ).
8.3.4 Basic simulation parameters
In Calculation type box, you can choose to perform Conjugate
gradients minimization, Velocity quenching minimization (not
recommended) and Molecular dynamics.
8.3.4.1 Timestep parameters
- Number of timesteps - numsteps
The number of simulation timesteps to be performed.
- Timestep size (fs) - timestep
The timestep size to use when integrating each step of the simulation. The
value is specified in femtoseconds.
- Starting timestep value - firsttimestep
The number of the first timestep. This value is typically used only when a
simulation is a continuation of a previous simulation. In this case, rather
than having the timestep restart at 0, a specific timestep number can be
specified.
- Timesteps per cycle - stepspercycle
Number of timesteps in each cycle. Each cycle represents the number of
timesteps between atom reassignments.
8.3.4.2 Multiple timestep parameters
- Timesteps between full elect. evaluations - fullElectFrequency
This parameter specifies the number of timesteps between each full
electrostatics evaluation.
- Timesteps between nonbonded evaluations - nonbondedFreq
This parameter specifies how often short-range non-bonded interactions
should be calculated. Setting it between 1 and Timesteps between full
elect. evaluations (fullElectFrequency) allows triple timestepping
where, for example, one could evaluate bonded forces every 1 fs, short-range
nonbonded forces every 2 fs, and long-range electrostatics every 4 fs.
- MTS algirithm type - MTSAlgorithm
Specifies the multiple timestep algorithm used to integrate the long and
short range forces. Impulse/VerletI is the same as r-RESPA and
Constant/Naive is the stale force extrapolation method.
- Long and short range forces split - longSplitting
Specifies the method used to split electrostatic forces between long and
short range potentials. It can be C1 (default) or Xplor. For
more details, see the NAMD
NAMD User Guide.
- Modified impulse method (MOLLY) - molly
This method eliminates the components of the long range electrostatic forces
which contribute to resonance along bonds to hydrogen atoms, allowing Timesteps
between full elect. evaluations (fullElectFrequency) of 6 (vs. 4) with a
1 fs timestep without using ShakeH type all (rigidBonds all). You may
use rigidBonds water but using ShakeH type all (rigidBonds all) with
MOLLY makes no sense since the degrees of freedom which MOLLY protects from
resonance are already frozen.
- Tolerance for MOLLY errors - mollyTolerance
Convergence criterion for MOLLY algorithm.
- Max. MOLLY iterations - mollyIterartions
Maximum number of iterations for MOLLY algorithm.
8.3.5 Input
In this page, you can set the input file name and the force field for the
simulation.
- Automatic input
Checking the Automatic input (default) the fields are automatically filled
using the molecule name in the current workspace.
- Force field
Force field that will be used in the simulation. The combo-box is
automatically updated when the dialog window is shown, reading the list of
the parameter files in the ...\VEGA ZZ\Data\ with the .inp
extension.
- Coordinate PDB file - coordinates
The PDB file containing initial position coordinate data.
- Binary PDB file - bincoordinates
PDB binary file used to restart the calculation.
- Parameter file - parameters
A parameter file defining all or part of the parameters necessary for
the molecular system to be simulated. You can specify more then one
parameter file separating them by a space character. The file requester
allows multiple selection.
- Format - paraTypeXplor, paraTypeCharmm
Specifies if the parameter file is in CHARMM or Xplor format.
- Velocities file - velocities, binvelocities
It's the PDB file containing the initial velocities for all atoms in the
simulation. This is typically a restart file or final velocity file written
by NAMD during a previous simulation.
- Binary velocities
If it's checked, the Velocities file is in binary format.
8.3.6 Output
This page allows to control the output parameters and the output file names.
- Output file - outputname
At the end of every simulation, NAMD writes two PDB files: the former
containing the final coordinates and the latter containing the final velocities of all
atoms in the simulation. This option specifies the file prefix for these two
files. The position coordinates will be saved to a file named as this prefix
with .coor appended. The velocities will be saved to a file named as
this prefix and with .vel suffix.
- Output frequency - outputEnergies
The number of timesteps between each energy output of NAMD. This value
specifies how often NAMD should output the current energy values. By
default, this is done every step. For long simulations, the amount of output
generated by NAMD can be greatly reduced by outputting the energies only
occasionally.
- Binary output - binaryoutput
Activates the use of binary output files. If this option is set to Yes,
then the final output files will be written in binary rather than PDB
format. Binary files preserve more accuracy between NAMD restarts than ASCII
PDB files, but the binary files are not guaranteed to be transportable
between computer architectures.
- Trajectory file - DCDfile
The binary DCD position coordinate trajectory filename. This file stores the
trajectory of all atom position coordinates using the same format (binary
DCD) as X-PLOR.
- Trajectory frequency - DCDfreq
The number of timesteps between the writing of position coordinates to the
trajectory file. The initial positions will not be included in the
trajectory file.
- Velocity trajectory file - velDCDfile
The binary DCD velocity trajectory filename. This file stores the trajectory
of all atom velocities using the same format (binary DCD) as X-PLOR.
- Velocity frequency - velDCDfreq
The number of timesteps between the writing of velocities to the trajectory
file. The initial velocities will not be included in the trajectory file.
- Restart file prefix - restartname
The prefix to use for restart filenames. NAMD produces PDB restart files
that store the current positions and velocities of all atoms at some step of
the simulation. This option specifies the prefix to use for restart files.
- Restart frequency - restartfreq
The number of timesteps between the generation of restart files.
- Binary restart - binaryrestart
Activates the use of binary restart files. If this option is checked, then
the restart files will be written in binary rather than PDB format. Binary
files preserve more accuracy between NAMD restarts than ASCII PDB files, but
the binary files are not guaranteed to be transportable between computer
architectures.
- Remove files after minimization
Checking it, all output files generated by minimization are removed. This
option has no effects if you are performing a MD simulation.
- Timestep in restart - restartsave
Appends the current timestep to the restart filename prefix, producing a
sequence of restart files rather than only the last version written.
- Write unit cell data - DCDUnitCell
If this option is set to Yes, then DCD files will contain unit cell
information in the style of CHARMM DCD files.
8.3.6.1 Output file parameters
- Add crossterm energy to dihedral - mergeCrossterms
If crossterm (or CMAP) terms are present in the potential, the energy is
added to the dihedral energy to avoid altering the energy output format.
- Momentum output frequency - outputMomenta
The number of timesteps between each momentum output of NAMD. If specified
and nonzero, linear and angular momenta will be printed to the output.
- Pressure output frequency - outputPressure
The number of timesteps between each pressure output of NAMD. If specified
and nonzero, atomic and group pressure tensors will be printed to the
output.
- Timing output frequency - outputTiming
The number of timesteps between each timing output of NAMD. If nonzero, CPU
and wallclock times and memory usage will be printed to the output.
8.3.7 Simulation space partitioning
- Cutoff (Å) - cutoff
Cutoff value for non-bond energy evaluation.
- Switching function - switching
If switching function is specified to be Off, then a truncated cutoff
is performed. If switching function is turned on, then smoothing functions
are applied to both the electrostatics and Van der Waals forces.
- Activation switching distance - switchdist
Distance at which the switching function should begin to take effect. This
parameter only has meaning if the switching function is set to On.
- Max. distance for limit interaction (Å) -
limitdist
The electrostatic and Van der Waals potential functions diverge as the
distance between two atoms approaches zero. The potential for atoms closer
than limitdist is instead treated as ar2 + c with
parameters chosen to match the force and potential at limitdist. This
option should primarily be useful for alchemical free energy perturbation
calculations, since it makes the process of creating and destroying atoms
far less drastic energetically. The larger the value of limitdist the
more the maximum force between atoms will be reduced. In order to not alter
the other interactions in the simulation, limitdist should be less
than the closest approach of any non-bonded pair of atoms; 1.3 Å appears to
satisfy this for typical simulations but the user is encouraged to
experiment. There should be no performance impact from enabling this
feature.
- Distance for inclusion in pair list (Å) -
pairlistdist
A pair list is generated pairlistsPerCycle times each cycle,
containing pairs of atoms for which electrostatics and Van der Waals
interactions will be calculated. This parameter is used when switching is
set to on to specify the allowable distance between atoms for inclusion in
the pair list. This parameter is equivalent to the X-PLOR parameter CUTNb.
If no atom moves more than pairlistdist - cutoff during one cycle,
then there will be no jump in electrostatic or Van der Waals energies when
the next pair list is built. Since such a jump is unavoidable when
truncation is used, this parameter may only be specified when switching is
set to on. If this parameter is not specified and switching is set to on,
the value of cutoff is used. A value of at least one greater than
cutoff is recommended.
- How to assign atoms to patches - splitpatch
When set to hydrogen, hydrogen atoms are kept on the same patch as
their parents, allowing faster distance checking and rigid bonds.
- Group-based distance testing (Å) - hgroupCutoff
This should be set to twice the largest distance which will ever occur
between a hydrogen atom and its mother. Warnings will be printed if this is
not the case. This value is also added to the margin.
- Extra length in patch dimension (Å) - margin
An internal tuning parameter used in determining the size of the cubes of
space with which NAMD uses to partition the system. The value of this
parameter will not change the physical results of the simulation. Unless you
are very motivated to get the very best possible performance, just leave
this value at the default.
- Min. procs for pairlist - pairlistMinProcs
Pairlists may consume a large amount of memory as atom counts, densities,
and cutoff distances increase. Since this data is distributed across
processors it is normally only problematic for small processor counts. Set
this parameter to the smallest number of processors on which the simulation
can fit into memory when pairlists are used.
- Regenerate x times per cycle - pairlistsPerCycle
Rather than only regenerating the pairlist at the beginning of a cycle,
regenerate multiple times in order to better balance the costs of atom
migration, pairlist generation, and larger pairlists.
- How often to print warnings - outputPairlists
If an atom moves further than the pairlist tolerance during a simulation
(initially (pairlistdist - cutoff)/2 but refined during the run) any
pairlists covering that atom are invalidated and temporary pairlists are
used until the next full pairlist regeneration. All interactions are
calculated correctly, but efficiency may be degraded. Enabling this
parameter will summarize these pairlist violation warnings periodically
during the run.
- Tolerance reduction fraction - pairlistShrink
In order to maintain validity for the pairlist for an entire cycle,
the pairlist tolerance (the distance an atom can move without causing the
pairlist to be invalidated) is adjusted during the simulation. Every time
pairlists are regenerated the tolerance is reduced by this fraction.
- Tolerance increasing fraction - pairlistGrow
In order to maintain validity for the pairlist for an entire cycle, the
pairlist tolerance (the distance an atom can move without causing the
pairlist to be invalidated) is adjusted during the simulation. Every time an
atom exceeds a trigger criterion that is some fraction of the tolerance
distance, the tolerance is increased by this fraction.
- Tolerance trigger - pairlistTrigger
The goal of pairlist tolerance adjustment is to make pairlist invalidations
rare while keeping the tolerance as small as possible for best performance.
Rather than monitoring the (very rare) case where atoms actually move more
than the tolerance distance, we reduce the trigger tolerance by this
fraction. The tolerance is increased whenever the trigger tolerance is
exceeded, as specified by pairlistGrow.
8.3.8 Dynamics
- Exclusion policy - exclude
This parameter specifies which pairs of bonded atoms should be excluded from
non-bonded interactions. With the value of none, no bonded pairs of
atoms will be excluded. With the value of 1-2, all atom pairs that
are directly connected via a linear bond will be excluded. With the value of
1-3, all 1-2 pairs will be excluded along with all pairs of atoms
that are bonded to a common third atom (i.e., if atom A is bonded to atom B
and atom B is bonded to atom C, then the atom pair A-C would be excluded).
With the value of 1-4, all 1-3 pairs will be excluded along with all
pairs connected by a set of two bonds (i.e., if atom A is bonded to atom B,
and atom B is bonded to atom C, and atom C is bonded to atom D, then the
atom pair A-D would be excluded). With the value of scaled1-4, all
1-3 pairs are excluded and all pairs that match the 1-4 criteria are
modified. The electrostatic interactions for such pairs are modified by the
constant factor defined by 1-4scaling. The van der Waals interactions
are modified by using the special 1-4 parameters defined in the parameter
files.
- Temperature (K) - temperature
Initial temperature value for the system. Using this option, a
random velocity distribution will be generated for the initial velocities for all the atoms
such that the system is at the desired temperature.
- Allow initial center of mass motion - COMmotion
Specifies whether or not motion of the center of mass of the entire system
is allowed. If this option is set to No, the initial velocities of
the system will be adjusted to remove center of mass motion of the system.
Note that this does not preclude later center-of-mass motion due to external
forces such as random noise in Langevin dynamics, boundary potentials, and
harmonic restraints.
- Remove center of mass drift for PME - zeroMomentum
If enabled, the net momentum of the simulation and any resultant drift is
removed before every full electrostatics step. This correction should
conserve energy and have minimal impact on parallel scaling. This feature
should only be used for simulations that would conserve momentum except for
the slight errors in PME (Features such as fixed atoms, harmonic restraints,
steering forces, and Langevin dynamics do not conserve momentum; use in
combination with these features should be considered experimental). Since
the momentum correction is delayed, enabling outputMomenta will show
a slight nonzero linear momentum but there should be no center of mass
drift.
- Dielectric constant - dielectric
Dielectric constant for the system. A value of 1.0 implies no modification
of the electrostatic interactions. Any larger value will lessen the
electrostatic forces acting in the system.
- Scaling factor for nonbonded forces - nonbondedScaling
Scaling factor for electrostatic and Van der Waals forces. A value of 1.0
implies no modification of the interactions. Any smaller value will lessen
the non-bonded forces acting in the system.
- Scaling factor for 1-4 interactions - 1-4scaling
Scaling factor for 1-4 interactions. This factor is only used when the
Exclusion policy (exclude) parameter is set to scaled1-4.
In this case, this factor is used to modify the electrostatic interactions
between 1-4 atom pairs. If the exclude parameter is set to anything but
scaled1-4, this parameter has no effect regardless of its value.
- Use geometric mean for L-J sigmas - vdwGeometricSigma
Use geometric mean, as required by OPLS, rather than traditional arithmetic
mean when combining Lennard-Jones sigma parameters for different atom types.
- Random number seed - seed
Number used to seed the random number generator if temperature or langevin
is selected. This can be used so that consecutive simulations produce the
same results. If no value is specified, NAMD will choose a pseudo-random
value based on the current UNIX clock time. The random number seed will be
output during the simulation startup so that its value is known and can be
reused for subsequent simulations. Note that if Langevin dynamics are used
in a parallel simulation (i.e., a simulation using more than one processor)
even using the same seed will not guarantee reproducible results.
- ShakeH type - rigidBonds
When water is selected, the hydrogen-oxygen and hydrogen-hydrogen
distances in waters are constrained to the nominal length or angle given in
the parameter file, making the molecules completely rigid. When this
parameter is all, waters are made rigid as described above and the bond
between each hydrogen and the (one) atom to which it is bonded is similarly
constrained. For the default case none, no lengths are constrained.
- Bond-length error for ShakeH (Å) -
rigidTolerance
The ShakeH algorithm is assumed to have converged when all constrained bonds
differ from the nominal bond length by less than this amount.
- Max. ShakeH iterations - rigidIterations
The maximum number of iterations ShakeH will perform before giving up on
constraining the bond lengths. If the bond lengths do not converge, a
warning message is printed, and the atoms are left at the final value
achieved by ShakeH. Although the default value is 100, convergence is
usually reached after fewer than 10 iterations.
- Exit if a ShakeH error occurs - rigidDieOnError
Exits and reports an error if rigidTolerance is not achieved after
rigidIterations.
- Use SETTLE for waters - useSETTLE
If rigidBonds are enabled then use the non-iterative SETTLE algorithm
to keep waters rigid rather than the slower SHAKE algorithm.
8.3.9 PME - Particle Mesh Ewald
PME stands for Particle Mesh Ewald and is an efficient full electrostatics
method for use with periodic boundary conditions. None of the parameters should
affect energy conservation, although they may affect the accuracy of the results
and momentum conservation.
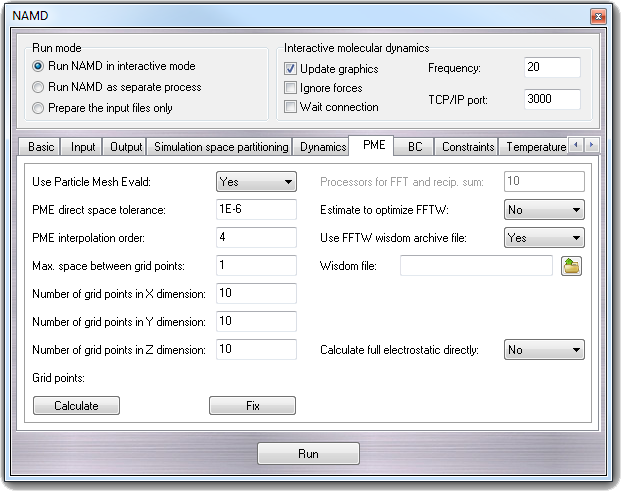
- Use Particle Mesh Evald - PME
Turns on Particle Mesh Ewald.
- PME direct space tolerance - PMETolerance
Affects the value of the Ewald coefficient and the overall accuracy of the
results.
- PME interpolation order - PMEInterpOrder
Charges are interpolated onto the grid and forces are interpolated off using
this many points, equal to the order of the interpolation function plus one.
- Max. space between grid points - PMEGridSpacing
The grid spacing partially determines the accuracy and efficiency of PME. If
any of the grid sizes below are not set, then PMEGridSpacing must be
set (recommended value is 1.0 Å) and will be used to calculate them. If a
grid size is set, then the grid spacing must be at least PMEGridSpacing
(if set, or a very large default of 1.5).
- Number of grid points in X dimension - PMEGridSizeX
Number of grid points in Y dimension - PMEGridSizeY
Number of grid points in Z dimension - PMEGridSizeZ
The grid size partially determines the accuracy and efficiency of PME. For
speed, the value should have only small integer factors (2, 3 and 5). To fix
values to be factors of 2, 3 and 5, you can click the Fix buttons. To
calculate automatically the grid points, you can click the Calculate
button.
- Processors for FFT and recip. sum - PMEProcessors
For best performance on some systems and machines, it may be necessary to
restrict the amount of parallelism used. Experiment with this parameter if
your parallel performance is poor when PME is used.
- Estimate to optimize FFTW - FFTWEstimate
Do not optimize FFT based on measurements, but on FFTW rules of thumb. This
reduces startup time, but may affect performance.
- Use FFTW wisdom archive fie - FFTWUseWisdom
Try to reduce startup time when possible by reading FFTW ``wisdom'' from a
file, and saving wisdom generated by performance measurements to the same
file for future use. This will reduce startup time when running the same
size PME grid on the same number of processors as a previous run using the
same file.
- Wisdom file - FFTWWisdomFile
File where FFTW wisdom is read and saved. If you only run on one platform
this may be useful to reduce startup times for all runs. The default is
likely sufficient, as it is version and platform specific.
- Calculate full electrostatic directly - FullDirect
Specifies whether or not direct computation of full electrostatics should be
performed.
8.3.10 BC - Boundary Conditions
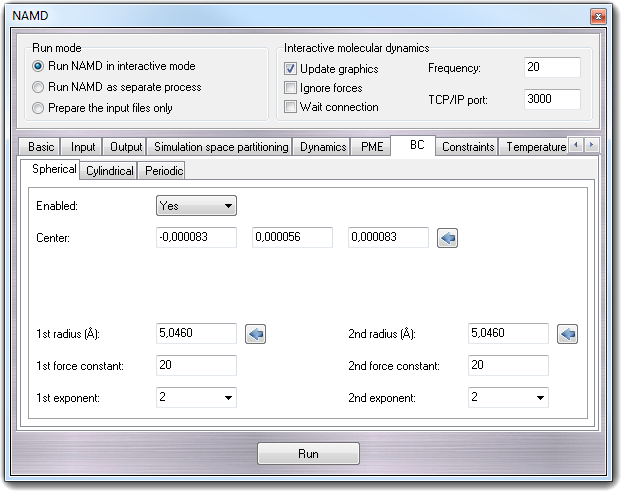
8.3.10.1 Spherical
NAMD provides spherical harmonic boundary conditions. These boundary
conditions can consist of a single potential or a combination of two potentials.
The following parameters are used to define these boundary conditions.
- Enabled - sphericalBC
Specifies whether or not spherical boundary conditions are to be applied to
the system.
- Center - sphericalBCCenter
Location around which sphere is cantered. Clicking the calculator button,
the fields are automatically filled with the center of mass of the molecule.
- 1st radius (Å) - sphericalBCr1
Distance at which the first potential of the boundary conditions takes
effect. This distance is a radius from the center. Clicking the calculator
button, the field is automatically filled with the the radius of the
hypothetical sphere containing the molecule.
- 1st force constant - sphericalBCk1
Force constant for the first harmonic potential. A positive value will push
atoms toward the center, and a negative value will pull atoms away from the
center.
- 1st exponent - sphericalBCexp1
Exponent for first boundary potential. The only likely values to use are 2
and 4.
- 2nd radius (Å) - sphericalBCr2
Distance at which the second potential of the boundary conditions takes
effect. This distance is a radius from the center. Clicking the calculator
button, the field is automatically filled with the the radius of the
hypothetical sphere containing the molecule.
- 2nd force
constant - sphericalBCk2
Force constant for the second harmonic potential. A positive value will push
atoms toward the center, and a negative value will pull atoms away from the
center.
- 2nd exponent -
sphericalBCexp2
Exponent for second boundary potential. The only likely values to use are 2
and 4.
8.3.10.2 Cylindrical
NAMD provides cylindrical harmonic boundary conditions. These boundary
conditions can consist of a single potential or a combination of two potentials.
The following parameters are used to define these boundary conditions.
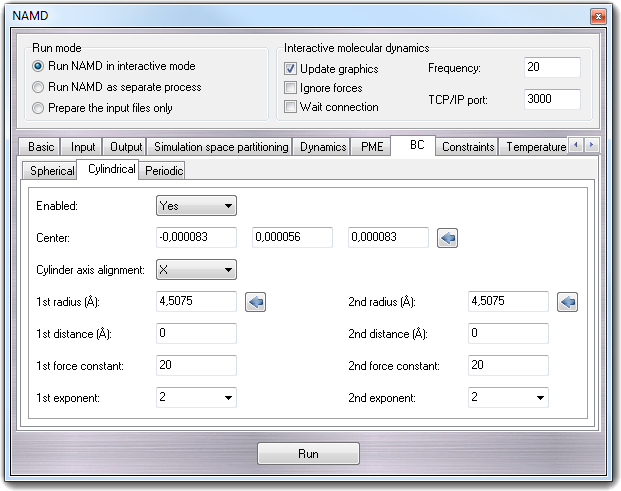
- Enabled - cylindricalBC
Specifies whether or not cylindrical boundary conditions are to be applied
to the system.
- Center - cylindricalBCCenter
Location around which cylinder is cantered. Clicking the calculator button,
the fields are automatically filled with the center of mass of the molecule.
- Cylinder axis alignment - cylinderBCAxis
Axis along which cylinder is aligned.
- 1st radius (Å) - cylindricalBCr1
Distance at which the first potential of the boundary conditions takes
effect along the non-axis plane of the cylinder. Clicking the calculator
button, the field is automatically filled with the the radius of the
hypothetical sphere containing the molecule.
- 1st distance (Å) - cylindricalBCl1
Distance at which the first potential of the boundary conditions takes
effect along the cylinder axis.
- 1st force constant - cylindricalBCk1
Force constant for the first harmonic potential. A positive value will push
atoms toward the center, and a negative value will pull atoms away from the
center.
- 1st exponent - cylindricalBCexp1
Exponent for first boundary potential. The only likely values to use are 2
and 4.
- 2nd radius (Å) - cylindricalBCr2
Distance at which the second potential of the boundary conditions takes
effect along the non-axis plane of the cylinder. Clicking the calculator
button, the field is automatically filled with the the radius of the
hypothetical sphere containing the molecule.
- 2st distance (Å)
- cylindricalBCl2
Distance at which the second potential of the boundary conditions takes
effect along the cylinder axis.
- 2nd force constant - cylindricalBCk2
Force constant for the second harmonic potential. A positive value will push
atoms toward the center, and a negative value will pull atoms away from the
center.
- 2nd exponent - cylindricalBCexp2
Exponent for second boundary potential. The only likely values to use are 2
and 4.
8.3.10.3 Periodic
NAMD provides periodic boundary conditions in 1, 2 or 3 dimensions. The
following parameters are used to define these boundary conditions.
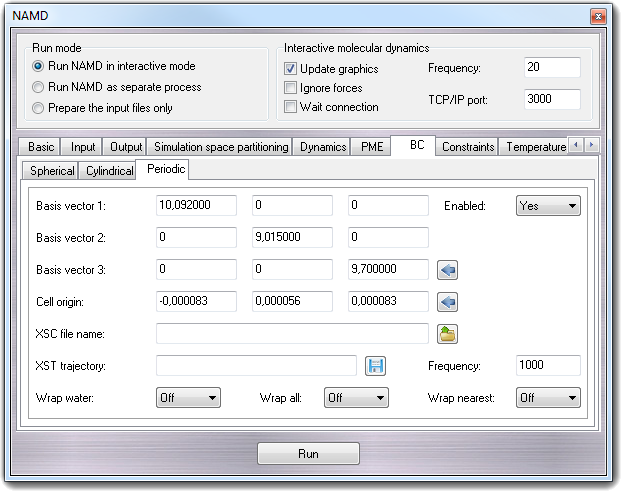
- Enabled
Enables the periodic boundary conditions.
- Basis vector 1 - cellBasisVector1
Basis vector 2 - cellBasisVector2
Basis vector 3 - cellBasisVector3
They specify the basis vectors for periodic boundary conditions. Clicking
the calculator button, all fields are filled with the cell dimensions of the
molecule.
- Cell origin - cellOrigin
When position rescaling is used to control pressure, this location will
remain constant. Also used as the center of the cell for wrapped output
coordinates. Clicking the calculator button, the fields are filled with the
mass center of the molecule.
- XSC file name - extendedSystem
In addition to .coor and .vel output files, NAMD generates a .xsc
(eXtended System Configuration) file which contains the periodic cell
parameters and extended system variables, such as the strain rate in
constant pressure simulations. Periodic cell parameters will be read from
this file if this option is present, ignoring the above parameters.
- XST trajectory - XSTfile
NAMD can also generate a .xst (eXtended System Trajectory) file which
contains a record of the periodic cell parameters and extended system
variables during the simulation.
- Frequency - XSTfreq
Controls how often the extended system configuration will be appended to the
XST file.
- Wrap water - wrapWater
Coordinates are normally output relative to the way they were read in.
Hence, if part of a molecule crosses a periodic boundary it is not
translated to the other side of the cell on output. This option alters this
behaviour for water molecules only.
- Wrap all - wrapAll
Coordinates are normally output relative to the way they were read in.
Hence, if part of a molecule crosses a periodic boundary it is not
translated to the other side of the cell on output. This option alters this
behavior for all contiguous clusters of bonded atoms.
- Wrap nearest - wrapNearest
Coordinates are normally wrapped to the diagonal unit cell centered on the
origin. This option, combined with wrapWater or wrapAll, wraps
coordinates to the nearest image to the origin, providing hexagonal or other
cell shapes.
8.3.11 Constraints
Before to proceed with a constrained simulation, the constraint constants must
be applied to the molecule. For more information, see the
Atom constraints section.
8.3.11.1 Harmonic constraints parameters
The following describes the parameters for the harmonic constraints feature
of NAMD. Actually, this feature should be referred to as harmonic restraints
rather than constraints, but for historical reasons the terminology of harmonic
constraints has been carried over from X-PLOR. This feature allows a harmonic
restraining force to be applied to any set of atoms in the simulation.
- Constraints - constraints
Specifies whether or not harmonic constraints are active. If it is set to
Off, then no harmonic constraints are computed.
- Constraint exponent - consexp
Exponent to be use in the harmonic constraint energy function.
- Constants file - conskfile
PDB file to use for force constants for harmonic constraints. By default,
this field is filled with the PDB file name including the atom coordinates
and generated automatically when the user presses the Run button.
- Ref. pos. file - consref
PDB file to use for reference positions for harmonic constraints. Each atom
that has an active constraint will be constrained about the position
specified in this file. By default, this field is filled with the PDB file
name including the atom coordinates and generated automatically when the
user presses the Run button.
- PDB column with the force constants - conskcol
Column of the PDB file to use for the harmonic constraint force constant.
This parameter may specify any of the floating point fields of the PDB file,
either X, Y, Z, occupancy, or beta-coupling
(temperature-coupling). Regardless of which column is used, a value of 0
indicates that the atom should not be constrained. Otherwise, the value
specified is used as the force constant for that atom's restraining
potential. The default value is Beta-coupling (B) because it's the
column used by VEGA ZZ to save the constraint constants.
- Scaling factor for energy function - constraintScaling
The harmonic constraint energy function is multiplied by this parameter,
making it possible to gradually turn off constraints during equilibration.
- Restraint the Cartesian coord. only - selectConstraints
This option is useful to restrain the positions of atoms to a plane or a
line in space. If active, this option will ensure that only selected
Cartesian components of the coordinates are restrained. E.g.: Restraining
the positions of atoms to their current z values with no restraints in X and
Y will allow the atoms to move in the X-Y plane while retaining their
original Z-coordinate. Restraining the X and Y values will lead to free
motion only along the Z coordinate.
- X - selectConstrX
Y - selectConstrY
Z - selectConstrZ
Restrain the Cartesian X, Y and Z components of the positions.
8.3.11.2 Fixed atoms parameters
Atoms may be held fixed during a simulation.
- Fixed atoms - fixedAtoms
Specifies whether or not fixed atoms are present.
- Calculate forces between fixed atoms - fixedAtomsForces
Specifies whether or not forces between fixed atoms are calculated. This
option is required to turn fixed atoms off in the middle of a simulation.
These forces will affect the pressure calculation, and you should leave this
option off when using constant pressure if the coordinates of the fixed
atoms have not been minimized. The use of constant pressure with significant
numbers of fixed atoms is not recommended.
- Fixed atoms file - fixedAtomsFile
PDB file to use for the fixed atom flags for each atom. If this parameter is
not specified, then the PDB file specified by coordinates is used. By
default, this field is filled with the PDB file name including the atom
coordinates and generated automatically when the user presses the Run
button.
- PDB column with fixed atoms - fixedAtomsCol
Column of the PDB file to use for the containing fixed atom parameters for
each atom. The coefficients can be read from any floating point column of
the PDB file. A value of 0 indicates that the atom is not fixed. Regardless
of which column is used, a value of 0 indicates that the atom should not be
constrained. Otherwise, the value specified is used as the force constant
for that atom's restraining potential. The default value is Beta-coupling
(B) because it's the column used by VEGA ZZ to save the constraint
constants.
8.3.12 Temperature
8.3.12.1 Langevin
NAMD is capable of performing Langevin dynamics, where additional damping and
random forces are introduced to the system. This capability is based on that
implemented in X-PLOR which is detailed in the X-PLOR User's Manual, although a
different integrator is used.
- Langevin dynamics - langevin
Specifies whether or not Langevin dynamics active.
- Temperature (K) - langevinTemp
Temperature to which atoms affected by Langevin dynamics will be adjusted.
This temperature will be roughly maintained across the affected atoms
through the addition of friction and random forces.
- Damping coefficient (1/ps) - langevinDamping
Langevin coupling coefficient to be applied to all atoms (unless
langevinHydrogen is off, in which case only non-hydrogen atoms are
affected). If not given, a PDB file is used to obtain coefficients for each
atom (see langevinFile and langevinCol below).
- Apply to hydrogens - langevinHydrogen
If this parameter is set to off then the Langevin dynamics the hydrogen
atoms aren't considered. This parameter has no effect if Langevin coupling
coefficients are read from a PDB file.
- Parameters - langevinFile
PDB file to use for the Langevin coupling coefficients for each atom. If
this parameter is not specified, then the PDB file specified by
coordinates is used.
- PDB column with coefficients - langevinCol
Column of the PDB file to use for the Langevin coupling coefficients for
each atom. The coefficients can be read from any floating point column of
the PDB file. A value of 0 indicates that the atom will remain unaffected.
8.3.12.2 Rescaling
NAMD allows equilibration of a system by means of temperature rescaling.
Using this method, all of the velocities in the system are periodically rescaled
so that the entire system is set to the desired temperature. The following
parameters specify how often and to what temperature this rescaling is
performed.
- Temperature rescaling frequency - rescaleFreq
The equilibration feature of NAMD is activated by specifying the number of
timesteps between each temperature rescaling. If this value is given, then
the rescaleTemp parameter must also be given to specify the target
temperature.
- Temperature for equilibration (K) - rescaleTemp
The temperature to which all velocities will be rescaled every
rescaleFreq timesteps.
8.3.12.3 Coupling
NAMD is capable of performing temperature coupling, in which forces are added
or reduced to simulate the coupling of the system to a heat bath of a specified
temperature. This capability is based on that implemented in X-PLOR which is
detailed in the X-PLOR User's Manual [7].
- Temperature coupling - tCouple
Specifies whether or not temperature coupling is active.
- Temperature (K) - tCoupleTemp
Temperature to which atoms affected by temperature coupling will be
adjusted. This temperature will be roughly maintained across the affected
atoms through the addition of forces.
- Parameters - tCoupleFile
PDB file to use for the temperature coupling coefficient for each atom. If
this parameter is not specified, then the PDB file specified by
coordinates is used.
- PDB column with coefficients - tCoupleCol
Column of the PDB file to use for the temperature coupling coefficient for
each atom. This value can be read from any floating point column of the PDB
file. A value of 0 indicates that the atom will remain unaffected.
8.3.14.4 Reassignment
NAMD allows equilibration of a system by means of temperature reassignment.
Using this method, all of the velocities in the system are periodically
reassigned so that the entire system is set to the desired temperature. The
following parameters specify how often and to what temperature this reassignment
is performed.
- Temperature reassignment frequency - reassingFreq
The equilibration feature of NAMD is activated by specifying the number of
timesteps between each temperature reassignment. If this value is given,
then the reassignTemp parameter must also be given to specify the
target temperature.
- Temperature for equilibration (K) - reassignTemp
The temperature to which all velocities will be reassigned every
reassignFreq timesteps. This parameter is valid only if reassignFreq
has been set.
- Temperature increment - reassignIncr
In order to allow simulated annealing or other slow heating/cooling
protocols, reassignIncr will be added to reassignTemp after
each reassignment (reassignment is carried out at the first timestep). The
reassignHold parameter may be set to limit the final temperature.
This parameter is valid only if reassignFreq has been set.
- Holding temperature - reassignHold
The final temperature for reassignment when reassignIncr is set;
reassignTemp will be held at this value once it has been reached. This
parameter is valid only if reassignIncr has been set.
8.3.13 Minimization
8.3.13.1 Conjugate gradients parameters
The default minimizer uses a sophisticated conjugate gradient and line search
algorithm with much better performance than the older velocity quenching method.
The method of conjugate gradients is used to select successive search directions
(starting with the initial gradient) which eliminate repeated minimization along
the same directions. Along each direction, a minimum is first bracketed
(rigorously bounded) and then converged upon by either a golden section search,
or, when possible, a quadratically convergent method using gradient information.
- First initial step for line minimizer - minTinyStep
If your minimization is immediately unstable, make this smaller.
- Max. initial step for line minimizer - minBabyStep
If your minimization becomes unstable later, make this smaller.
- Gradien reduction factor - minlLineGoal
Varying this might improve conjugate gradient performance.
8.3.13.2 Velocity quenching parameters
You can perform energy minimization using a simple quenching scheme. While
this algorithm is not the most rapidly convergent, it is sufficient for most
applications. There are only two parameters for minimization: one to activate
minimization and another to specify the maximum movement of any atom.
- Max. atom move (Å) - maximumMove
Maximum distance that an atom can move during any single timestep of
minimization. This is to insure that atoms do not go flying off into space
during the first few timesteps when the largest energy conflicts are
resolved.
8.3.14 Other
In Other parameters box, you can put parameters not managed by graphic interface and TCL scripts. The Configurations box includes the
gadgets to control the pre-settings: to configure NAMD, several commands can be
involved and thus it could be useful to save the configuration more then one
time. As explained in NAMD interface basics
section, you can Load, Save and revert to Default
configuration, pressing the buttons in the Configurations box or using the
context menu. The Ignore file name checkbox has the same function of the
homonymous item in the context menu: if it's checked, the fields containing file
names aren't updated when the configuration is loaded, in order to preserve the
names of the current molecule.
In the Presettings box, are shown the NAMD configuration files saved in
the ...\VEGA ZZ\Data\NAMD directory with the .namd extension. They
can be managed by the context menu displayed when you click with the right mouse
button on a presetting in the list.
| Menu item |
Description |
| Load |
Load the preset into the graphic interface |
| Save |
Save the preset to a new file. |
| Update |
Update the current preset with the new changes. |
| Edit |
Edit the preset in text mode (see the
Mini Text Editor). |
| Rename |
Rename the preset. |
| Delete |
Delete the preset. A warning message is shown before
proceeding. |
| Refresh list |
Refresh the preset list. This function is useful if
you added manually al preset file in the ...\VEGA ZZ\Data\NAMD
directory and you want see it in the list without restarting VEGA ZZ. |
8.3.15 Starting the simulation
Clicking Run button, the simulation starts: the PDB file is
created with constraints parameters if required, the PSF topology file is
built and the NAMD command file as generated by the data fitted in the graphic
interface fields. All files are saved in the directory in which the current
molecule is loaded or saved.
Before to run NAMD, VEGA ZZ performs an extra check to verify if all parameters
are included in the force field data (the files are placed in ...\VEGA ZZ\Data\Parameters
directory). It may be possible, especially for the small molecules, that certain
parameters (bonds, angles, torsions, impropers) are missing and the
Missing parameter table is shown. Thanks to this
table, you can put the required parameters.