Molecular dynamics of a non-peptidic molecule with NAMD
1. Introduction
2. What's you need
3. Ibuprofen - water preparation
4. PSF file creation
5. NAMD minimization
6. Heating
7. Molecular dynamics
8. How to make resident the new parameter file
This tutorial explains how to perform the molecular dynamics of the ibuprofen - water system with NAMD. The ibuprofen is a non-peptidic molecule and so some potential parameters are missing in the standard CHARMM 22 force field. TYou can learn how to fix the missing force field parameters and how to run NAMD without the VEGA ZZ graphic interface.
3. Ibuprofen - water preparation
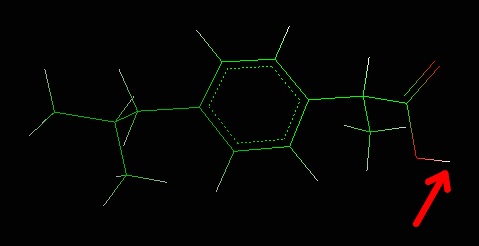
The hydrogen is now deleted.
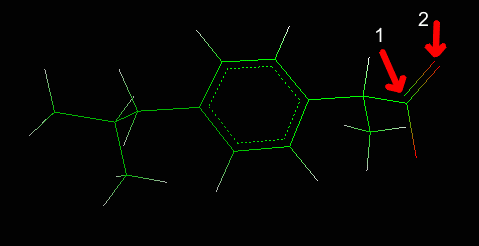
The bond will be changed from double to partial double.
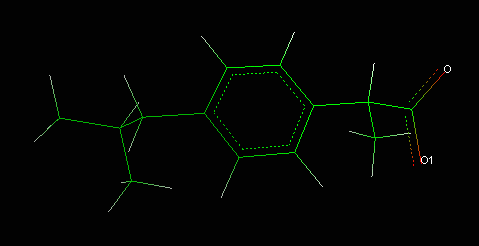
The ibuprofen doesn't have a standard topology and so the PSF file creation isn't totally automatic because some parameters aren't included in the CHARMM22_PROT parameter file. For more details about the input files required by NAMD, see this tutorial.
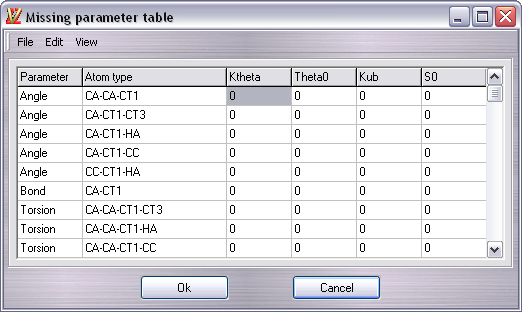
It indicates all angles, bonds and torsions parameters not included in the force field. In this window you can put manually the parameters, but if you don't know them, you can ask to the program to complete them for you.
Click Edit -> Auto assign and the table will be filled. Please remember that this isn't a full exaustive approach and it can generate wrong parameters. To check them, you can click on the missing parameter in the table and the involved atoms are automatically highlighted in the workspace.
Before to start the molecular dynamics simulation, an energy minimization is required.
numsteps 5000 minimization on dielectric 1.0coordinates ibuprofen_wat.pdb outputname ibuprofen_wat_min outputEnergies 1000 binaryoutput no DCDFreq 1000 restartFreq 1000structure ibuprofen_wat.psf paraTypeCharmm on parameters par_all22_prot.inp parameters par_all22_vega.inp parameters ibuprofen_wat.inp exclude scaled1-4 1-4scaling 1.0 switching on switchdist 8.0 cutoff 12.0 pairlistdist 13.5 margin 0.0 stepspercycle 20Save the file with the ibuprofen_wat_min.namd name. This performs a 5000 steps conjugate gradients minimization, saving the output (coordinates and restart files) every 1000 iterations. For more information about the parameters, please consult the NAMD User Guide.
namd2 ibuprofen_wat_min.namd > ibuprofen_wat_min.outand hit return. After few time the minimization is finish.
This is the first molecular dynamics phase required to set the atom velocities at the specified temperature.
numsteps 3000 dielectric 1.0coordinates ibuprofen_wat_min.pdb temperature 0 seed 12345outputname ibuprofen_wat_heat outputEnergies 1000 binaryoutput yes DCDFreq 1000 restartFreq 1000timestep 1.0 nonbondedFreq 1 fullElectFrequency 1structure ibuprofen_wat.psf paraTypeCharmm on parameters par_all22_prot.inp parameters par_all22_vega.inp parameters ibuprofen_wat.inp exclude scaled1-4 1-4scaling 1.0 switching on switchdist 8.0 cutoff 12.0 pairlistdist 13.5 margin 0.0 stepspercycle 20langevin on langevinDamping 10 langevinTemp 300 langevinHydrogen nosphericalBC on sphericalBCCenter 0.0 0.0 0.0 sphericalBCr1 16.00 sphericalBCk1 2.00
namd2 ibuprofen_wat_heat.namd > ibuprofen_wat_heat.outand hit return.
At the heating end, you must prepare the MD input file.
firsttimestep 3000 numsteps 103000 dielectric 1.0coordinates ibuprofen_wat_min.pdb bincoordinates ibuprofen_wat_heat.coor binvelocities ibuprofen_wat_heat.vel extendedsystem ibuprofen_wat_heat.xsc seed 12345outputname ibuprofen_wat_dyn outputEnergies 1000 binaryoutput yes DCDFreq 1000 restartFreq 1000timestep 1.0 nonbondedFreq 1 fullElectFrequency 1structure ibuprofen_wat.psf paraTypeCharmm on parameters par_all22_prot.inp parameters par_all22_vega.inp parameters ibuprofen_wat.inp exclude scaled1-4 1-4scaling 1.0 switching on switchdist 8.0 cutoff 12.0 pairlistdist 13.5 margin 0.0 stepspercycle 20langevin on langevinDamping 10 langevinTemp 300 langevinHydrogen nosphericalBC on sphericalBCCenter 0.0 0.0 0.0 sphericalBCr1 16.00 sphericalBCk1 2.00
namd2 ibuprofen_wat_dyn.namd > ibuprofen_wat_dyn.outand hit return.
8. How to make resident the new parameter file
If you don't want reassign the parameters every time that you want perform the ibuprofen NAMD calculation, you can store them in VEGA ZZ.
* * CHARMM 22 parameters file for VEGA ZZ * INCLUDE "Parameters/par_all22_lipid.inp" INCLUDE "Parameters/par_all22_na.inp" INCLUDE "Parameters/par_all22_prot.inp" INCLUDE "Parameters/par_all22_vega.inp" INCLUDE "Parameters/par_all22_user.inp"
* * CHARMM 22 parameters file for VEGA ZZ * INCLUDE "Parameters/par_all22_lipid.inp" INCLUDE "Parameters/par_all22_na.inp" INCLUDE "Parameters/par_all22_prot.inp" INCLUDE "Parameters/par_all22_vega.inp" INCLUDE "Parameters/par_all22_user.inp" INCLUDE "Parameters/ibuprofen_wat.inp"
A better procedure, is to merge all user-defined parameters in the par_all22_user.inp that must be placed in the ...\VEGA ZZ\Data\Parameters directory. In this way, you mustn't edit the CHARMM22_PROT.inp file in the ...\VEGA ZZ\Data directory.
inpmerge "...\VEGA ZZ\Data\Parameters\par_all22_user.inp" "...\MyPath\ibuprofen_wat.inp"where ...\VEGA ZZ is the VEGA ZZ installation path and the ...\MyPath is the full path of the ibuprofen_wat.inp file. For more information about the inpmerge utility, see its manual.
WARNING:
please remember that the INCLUDE command isn't implemented in NAMD and CHARMM,
because they don't have a macro pre-processor. This is a feature available in
VEGA ZZ only.