How to build a small molecule
1. Introduction
2. What's you need
3. The molecule to build
3.1 PubChem download
3.2 IUPAC builder
3.3 SMILES builder
3.4 Ketcher 2D editor
3.5 3D molecular editor
4. Structure optimization
5. Conformational search
6. Final optimization
1. Introduction
This tutorial explains how to build a small molecule (e.g. a ligand) by using
the 2D and 3D molecular editors included in VEGA ZZ and how to perform a systematic
conformational search (grid scan) in order to find the best conformer.
2. What you need
- VEGA ZZ release 3.0.2 or greater (click here for the
software setup).
- MOPAC 2012 (optional, click here
to know how to install it)
3. The molecule to build
VEGA ZZ includes some molecular editors with different features. The most
intuitive editors are 2D, because you can build a molecule as shown on a paper
sheet, but the most powerful is 3D, because allows you to change the molecules
directly in the workspace with a total control of stereochemistry and
geometrical isomerism. Moreover, VEGA ZZ can build molecules starting from their
1D structures from SMILES strings or IUPAC names, but these editors are not so
intuitive. Finally, if you are lucky, you can download the 3D structure from
PubChem database
and show it in the VEGA ZZ environment.
For example, imagine you want to build imipramine or
3-(10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-N,N-dimethylpropan-1-amine:
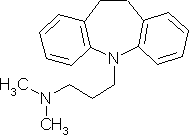
Imipramine
3.1 PubChem download
The easiest way to obtain a small molecule is to download it from PubChem
library. VEGA ZZ includes the possibility to do that without the use of a Web
browser.
- Select File
 Download
From PubChem and put imipramine in Molecule name to download.
Download
From PubChem and put imipramine in Molecule name to download.
- Click Ok and the molecule will be downloaded from PubChem and
shown in the current workspace.
3.2 IUPAC builder
If you know the 1D structure as IUPAC name of
a small molecule, you can try to build it directly by using this information.
- Select Edit
Build
IUPAC and IUPAC window will be shown.
- Put
3-(10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)-N,N-dimethylpropan-1-amine
in the IUPAC name field and click Build.
- The molecule will be converted to 3D and shown in the current workspace.
3.3 SMILES builder
Another possibility to build a molecule from 1D structure is the use of its
SMILES string.
- Select Edit
Build
SMILES and SMILES window will be shown.
- Put CN(C)CCCN1C2=CC=CC=C2CCC3=CC=CC=C31 in SMILES field and the 2D
preview will be automatically shown to allow you the correctness of the
string.
- Click Build and the molecule will be converted to 3D and shown in
the current workspace.
3.4 Ketcher 2D editor
VEGA ZZ package includes a 2D molecular editor named
Ketcher
provided by GGA Software Services. It's very intuitive and easy to use.
- Select Edit
Ketcher and the editor window will be shown.
- To build imipramine, start adding the cycloheptane ring by selecting it
in the menu which is shown by clicking two times (a long double click) with
the left mouse button on the icon shown as a benzene ring. Then, place the
ring at center on the Ketcher edit area by clicking with the left mouse
button.
- Select benzene as above and add the two benzene rings by clicking
the cycloheptane bonds.
- Change the order (from single to double) of both bonds shared by benzene
rings and cycloheptane in order to revert the aromaticity. To do it, click
the two bonds and the bond order will be changed from single to double.
- Click N in the element bar at the right in Ketcher window and click the
carbon 1 of the cycloheptane to change it to nitrogen.
- Add the n-butyl chain to the nitrogen by clicking the straight line of
the tool bar on the left and the nitrogen. A single methyl will be added.
Click the carbon of this methyl to add a second one and so on to obtain the
n.-butyl chain.
- As above, change the last carbon of the n-butyl chain to nitrogen and
add two methyl groups to it.
- The 2D structure is now finished. Click Send to VEGA ZZ to build the 3D
structure.
3.1 3D molecular editor
Imagine you want to build imipramine using the 3D molecular editor. The editor is based on fragment databases containing building blocks that
can be combined each other to complete a more complex structure. For this
reason, you must cut the molecule in less complex fragments that will be
assembled as indicated in the following scheme:
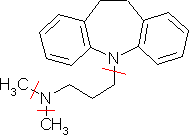
Excluding initially the heteroatoms, the molecule can be fragmented in a tricyclic system (Dibenzo[a,d]cycloheptane), in a n-butyl chain and in
two methyl groups.
- Select Edit
Add
Fragments in
main menu and
Add fragment/s window will appear.
- Choose Rings (aromatic) in Group box and
15 5H-Dibenzo[a,d]cycloheptane in
Fragment box.
- Click Finish and thus Close.
The starting building block is shown in the workspace:
- In order to change the C5 carbon to a nitrogen and to remove one hydrogen
connected to it, select Edit
Change
Atom/residue/chain in menu bar.
- Click C5 as indicated by the red arrow in the following picture:
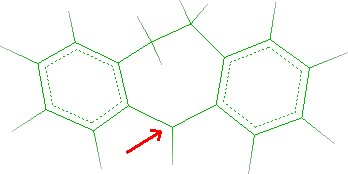
- In Element field of Edit dialog, change C to
N, click Apply and close the window. The atom color
will change from green to blue.
- Rotating the molecule, highlight the two hydrogens bonded to N5.
- To remove one hydrogen, select Edit
Remove
Atom and click the atom to remove as indicated by the red
arrow:
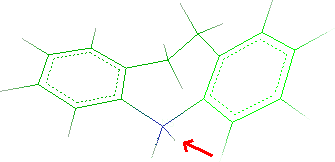
The hydrogen will be deleted. Click Done to close the
window.
- To add the n-butyl chain, reopen the Add fragment/s window (Edit
Add
Fragments), search 04C n-Butane
in the Alkanes group and click Next button.
- Click the butane hydrogen that will be merged with the hydrogen bonded to
N5 (see red arrow):
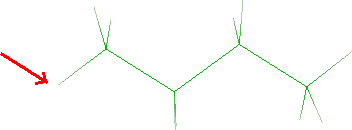
- Click Next button and the tricyclic system will be shown. Click
the hydrogen bonded to N5:
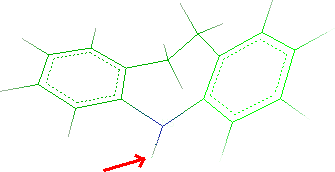
- Click the Next and thus Finish. Close the
window.
- As previously explained, you must change the C4 of the n-butyl
chain to a nitrogen and remove a hydrogen: select
Edit
Change
Atom/residue/chain and click the C4 as indicated by
the red arrow in the picture:
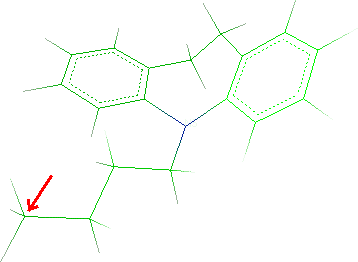
- In the Element field of the Edit dialog, change C to
N, click the Apply button and close the window.
- To remove one hydrogen, select Edit
Remove
Atom and click the atom to remove:
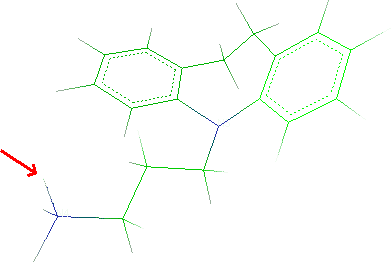
Click Done to close the window.
- Finally you must add two methyl groups to N4 of the n-butyl
chain. Open the Add fragment/s window (Edit
Add
Fragments).
- In Alkanes group select 01C Methane and click
Next.
- Click a methane hydrogen and Next button.
- Click a hydrogen of the ammine:
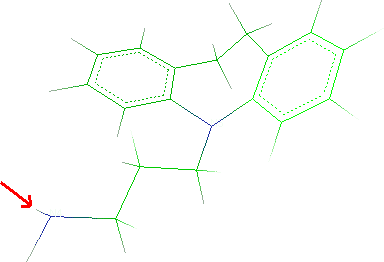
- Click Next and Finish.
- Repeat the same steps to add the second methyl group:
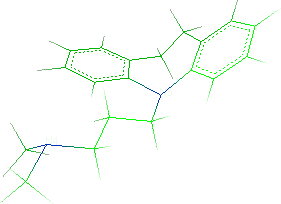
- Save the molecule in IFF format (File
Save
As...) with imipramine.iff name.
4. Structure optimization
In this section will be explained how to perform a conjugate gradient
minimization in order to optimize the rough 3D structure.
- Fix the atom types and the charges (Calculate
Charge & Pot.), checking Force field and Charges and selecting
SP4 and Gasteiger. Click Fix button. The total charge
is 0.
- Open the Ammp minimization
window selecting Calculate
Ammp
Minimization in the menu bar.
- Choose Conjugate gradients and set Minimization steps to
1000 and Toler to 0.01.
- Click Run button. After few steps, the minimization is
completed.
- Save the molecule in IFF format overwriting the previous one.
5. Conformational search
In order to find a reasonable lowest energy conformation, it will be
explained how to perform the conformational search of the built molecule. The
flexible torsions (dihedrals) will be systematically rotated by an angle value (grid
scan) and each conformation will be optimized in order to find the
best minimum.
- Open Ammp conformational search
window (Calculate
Ammp
Conformational search).
- Click Edit torsion button. The Selection
tool window will appear.
- In its menu bar, select Edit
Add flexible torsions and all
flexible torsion are automatically added in the selection list (4 torsions).
They are highlighted in the workspace also.
- Click Done button and the Ammp conformational search window is
shown.
- Select Systematic in Method field of Search
parameters box. The upper box shows the torsions that will be rotated during the scan.
Increasing the number of rotation steps (see the Steps field in
Torsion parameters box), the search is more accurate but more
computational time is required. The six value is a good choice because it
means a rotation 60 degrees of each step that in some cases is the threshold
to classify different conformations.
- Check Minimize all conformations and put 100 in the Steps
field and 0.01 in the Toler field. In this way, each conformation is
optimized using the conjugate gradients method. The minimization
finishes when the specified number of iterations (Steps) is reached or
the Toler condition is satisfied.
- If you want to analyze all conformations generated by the systematic search,
check Trajectory, Output and Energy in the left box.
Please remember to set the Graphic update to 1, otherwise not all
conformations will be stored in the output files. If you don't save the outputs,
only the lowest energy conformation is kept at the end of the calculation.
- Click Run button to start the calculation. In the console is
reported for each conformation the starting energy, the best energy found at
that time, and the energy after the minimization.
- After few time, the
search is finish and the best conformation is automatically loaded in the
workspace.
- To refine this structure, repeat the energy minimization as explained in
the previous section and save the final molecule.
6. Final optimization
If the molecular mechanics is not enough for you, you can perform a final
optimization by semi-empirical calculation such as MOPAC.
- Select Calculate
Mopac and the MOPAC dialog window will be shown.
- Click the Default button.
- In the Parameters box, choose PM7 as calculation type if
you have installed MOPAC 2012, otherwise choose PM1.
- Leave the other parameters to default and click Run.
- When the calculation is finished, the optimized structure is
automatically loaded and you can save it in your preferred file format.